Navigating FDA Guidelines for Nanotechnology Drug Development: A Strategic Framework for Industry Success
This article provides a comprehensive roadmap for researchers and drug development professionals engaging with the FDA on nanotechnology-based products.
Navigating FDA Guidelines for Nanotechnology Drug Development: A Strategic Framework for Industry Success
Abstract
This article provides a comprehensive roadmap for researchers and drug development professionals engaging with the FDA on nanotechnology-based products. It covers foundational regulatory concepts, practical methodologies for development and characterization, strategies for addressing common regulatory and technical challenges, and approaches for comparative analysis and final validation. The content synthesizes current FDA guidance and industry best practices to equip teams with the knowledge needed for successful pre-submission meetings and regulatory submissions.
Understanding the FDA's Framework for Nanotechnology Product Regulation
Nanotechnology involves the understanding and control of matter at dimensions between approximately 1 and 100 nanometers. The FDA's regulatory approach is based on a product-specific, science-based assessment. The central guidance is the 2014 final guidance document: "Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology." This document outlines the FDA's working definition, stating that a material may be considered to involve nanotechnology if it:
- Is engineered to have at least one external dimension, or an internal or surface structure, in the nanoscale range (approximately 1 nm to 100 nm), OR
- Is engineered to exhibit properties or phenomena, including physical or chemical properties or biological effects, that are attributable to its dimension(s), even if these dimensions fall outside the nanoscale range, up to one micrometer.
This broad, "case-by-case" definition underscores the FDA's focus on the properties and effects of a material, rather than a strict size cutoff.
Key FDA Guidance Documents and Guidelines
The FDA has issued several product-specific guidance documents to aid developers. The most current and relevant documents are summarized in the table below.
Table 1: Key FDA Nanotechnology Guidance Documents (as of 2024)
| Guidance Document Title (Year) | Product Center | Status & Purpose | Key Quantitative Recommendations/Considerations |
|---|---|---|---|
| Considering Whether an FDA-Regulated Product Involves the Nanotechnology (2014) | OC, CDER, CBER, CDRH, CFSAN, CVM | Final Guidance. Provides the FDA's working definition and framework for assessment. | Size range (1-100 nm). Evaluation of dimension-dependent properties up to 1 µm. |
| Drug Products, Including Biological Products, that Contain Nanomaterials (2022) | CDER, CBER | Final Guidance. Covers chemistry, manufacturing, and controls (CMC), safety, and efficacy for human drugs. | Recommends comprehensive physicochemical characterization (size, distribution, morphology, surface charge). Stability studies must monitor potential changes in nanomaterial properties. |
| Use of Nanomaterials in Food for Animals (2015) | CVM | Final Guidance. For food additives and GRAS substances in animal food. | Recommends safety assessments that account for altered ADME (Absorption, Distribution, Metabolism, Excretion). |
| Safety of Nanomaterials in Cosmetic Products (2014, updated 2022) | CFSAN | Guidance for Industry. Outlines safety considerations for manufacturers. | Recommends assessing penetration, reactivity, and systemic exposure. Particle size and aggregation state are critical parameters. |
| Assessing the Effects of Significant Manufacturing Process Changes... (2019) | CDER, CBER | Final Guidance. Includes considerations for nanomaterials. | For changes to nanomaterial manufacturing, bioequivalence or comparability studies may be needed if physicochemical changes impact product performance. |
| Final Guidance for Industry: Liposome Drug Products (2018) | CDER | Final Guidance. While not exclusively nano, liposomes are a key nanotechnology platform. | Specific recommendations for particle size distribution, lamellarity, drug release, and in vivo stability testing. |
Abbreviations: OC (Office of the Commissioner), CDER (Center for Drug Evaluation and Research), CBER (Center for Biologics Evaluation and Research), CDRH (Center for Devices and Radiological Health), CFSAN (Center for Food Safety and Applied Nutrition), CVM (Center for Veterinary Medicine).
Experimental Protocols: Key Characterization Assays for Regulatory Submission
This section provides detailed methodologies for essential characterization experiments referenced in FDA guidance.
Protocol 3.1: Comprehensive Physicochemical Characterization of Engineered Nanomaterials (EMN)
Objective: To determine the critical physicochemical attributes of an ENM as recommended in the 2022 guidance for drug products.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Sample Preparation: Prepare a representative sample (≥ 5 mg) in its relevant biological or vehicle matrix (e.g., PBS, cell culture medium, simulated gastric fluid) at the intended use concentration. Sonicate using a probe sonicator (70% amplitude, 5 cycles of 30 sec on/30 sec off on ice) to ensure dispersion.
- Dynamic Light Scattering (DLS) for Hydrodynamic Size & PDI:
- Load 1 mL of prepared sample into a low-volume disposable sizing cuvette.
- Equilibrate to 25°C for 300 seconds in the instrument.
- Perform measurement with a minimum of 12 sub-runs. Repeat for n=3 independent samples.
- Record the Z-average hydrodynamic diameter (d.nm) and the polydispersity index (PDI).
- Laser Diffraction for Particle Size Distribution (PSD):
- Use a laser diffraction instrument capable of measuring from 10 nm to 1000 µm.
- Add sample dropwise to the instrument's circulating background medium under agitation until an obscuration rate of 8-12% is achieved.
- Perform measurement (n=5). Report the volume-weighted D10, D50, D90 values and span [(D90-D10)/D50].
- Electron Microscopy for Primary Particle Size and Morphology:
- TEM Sample Prep: Dilute sample 1:100 in deionized water. Apply 5 µL to a carbon-coated copper grid. Blot dry after 60 seconds. Optionally, negative stain with 1% uranyl acetate.
- SEM Sample Prep: Deposit sample on a silicon wafer. Sputter-coat with 5 nm of gold/palladium.
- Imaging: Acquire micrographs at ≥ 50,000x magnification. Measure the primary particle diameter or longest dimension for n ≥ 200 particles using image analysis software.
- Zeta Potential Measurement:
- Load 800 µL of sample into a clear disposable zeta cell.
- Set instrument to 25°C and use the Smoluchowski model.
- Perform measurement with a minimum of 30 runs. Report the average zeta potential in millivolts (mV) (n=3).
Protocol 3.2:In VitroDrug Release Kinetics for Nanocarrier Formulations
Objective: To assess the drug release profile from a nanocarrier under sink conditions, critical for quality control and in vivo performance prediction.
Materials: See "The Scientist's Toolkit." Procedure:
- Dialysis Method:
- Place a volume of nanocarrier formulation containing 1-5 mg of drug into a pre-soaked dialysis cassette or tubing (MWCO ≤ 1/5th the size of the nanocarrier).
- Immerse the sealed dialysis device in 500 mL of release medium (e.g., PBS pH 7.4, with 0.5% w/v SDS to maintain sink conditions). Maintain medium at 37°C with constant stirring (100 rpm).
- Sampling: At predetermined time points (e.g., 0.5, 1, 2, 4, 8, 12, 24, 48, 72 h), withdraw 1 mL from the external medium and replace with an equal volume of fresh, pre-warmed release medium.
- Analysis: Quantify drug concentration in samples using a validated HPLC or UV-Vis method. Correct for sample replacement.
- Data Analysis: Calculate cumulative drug release (%) and fit data to release kinetic models (e.g., zero-order, first-order, Higuchi, Korsmeyer-Peppas).
Protocol 3.3: Endotoxin Testing for Parenteral Nanoformulations (LAL Assay)
Objective: To ensure parenteral nanoformulations meet USP endotoxin limits (< 5.0 EU/kg/hour for most drugs), as endotoxin can cause immune effects confounding nanomaterial safety studies.
Materials: Limulus Amebocyte Lysate (LAL) reagent, control standard endotoxin (CSE), endotoxin-free water and supplies. Procedure:
- Sample Preparation: Dilute the nanoformulation with endotoxin-free water to a concentration that falls within the validated range of the LAL assay (typically 0.05-0.2 EU/mL) and that is below the Maximum Valid Dilution (MVD).
- Spike Recovery (Validation): Prepare a parallel sample spiked with a known concentration of CSE (e.g., 0.1 EU/mL) to confirm the sample does not interfere with the assay (recovery must be 50-200%).
- Assay Execution (Kinetic Turbidimetric Method):
- Pre-incubate microplate with samples, standards (0.01-1.0 EU/mL), and controls at 37°C for 5 min in a plate reader.
- Add an equal volume of LAL reagent to each well.
- Immediately begin kinetic reading, monitoring absorbance at 340 nm every 30 seconds for 90 minutes.
- Determine the reaction time for each well.
- Calculation: Generate a standard curve (log reaction time vs. log endotoxin concentration). Interpolate the endotoxin concentration of the test sample and multiply by the dilution factor to report EU/mL of the original formulation.
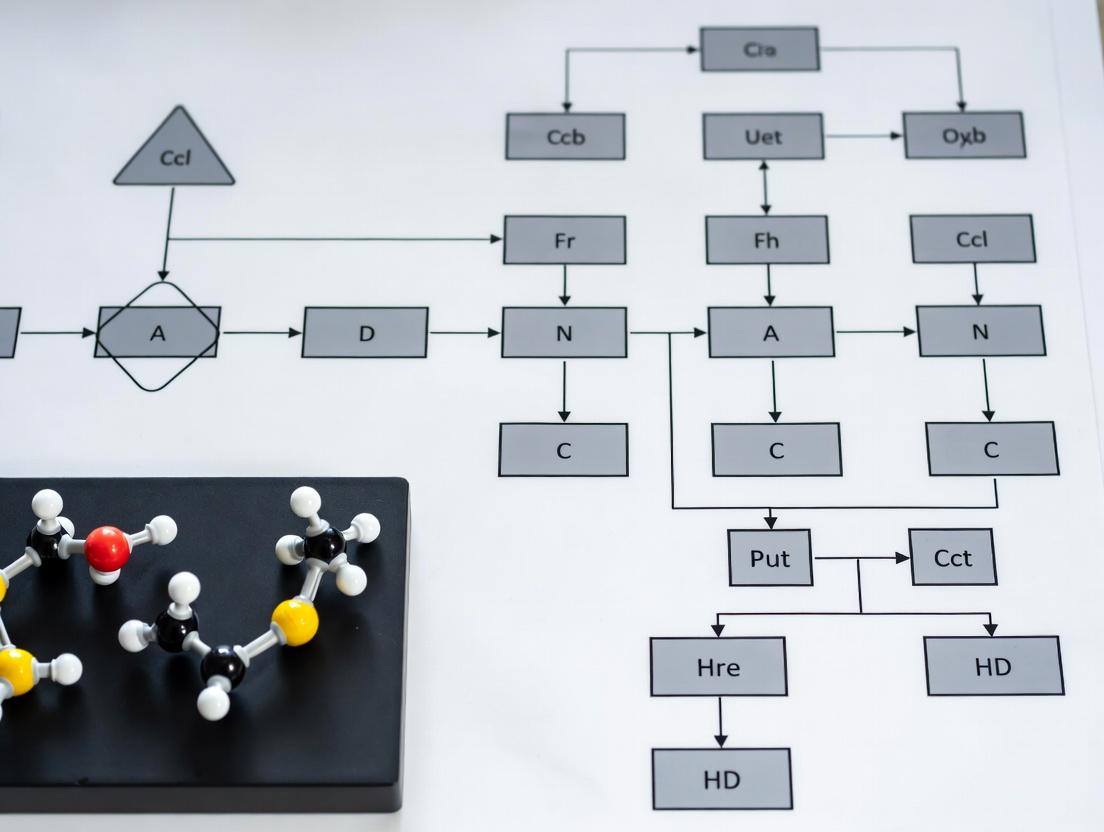
Visualizations
FDA Nanoproduct Assessment Workflow
Diagram Title: FDA Nanomaterial Product Assessment Decision Tree
Key Physicochemical Characterization Data Flow
Diagram Title: Key Nano-Characterization Assays and Outputs
The Scientist's Toolkit
Table 2: Essential Research Reagents & Materials for Regulatory Nano-Characterization
| Item/Category | Specific Example(s) | Function in Protocol |
|---|---|---|
| Size & Distribution Analysis | Dynamic Light Scattering (DLS) Instrument (e.g., Malvern Zetasizer); Laser Diffraction Analyzer (e.g., Beckman Coulter LS 13 320). | Measures hydrodynamic diameter, PDI (DLS) and broad particle size distribution (LD) per ICH Q2 guidelines. |
| Morphology & Primary Size | Transmission Electron Microscope (TEM); Scanning Electron Microscope (SEM); Carbon-coated copper grids; Silicon wafers. | Provides direct visualization and measurement of primary particle size, shape, and aggregation state. |
| Surface Charge Analysis | Zeta Potential Analyzer (often integrated with DLS); Disposable folded capillary cells. | Determines the electrostatic surface potential, predicting colloidal stability and interaction with biological components. |
| Drug Release & Stability | Dialysis cassettes/tubing (appropriate MWCO); USP-compliant dissolution apparatus; HPLC-UV system. | Quantifies drug release kinetics under controlled conditions to establish product performance and stability. |
| Endotoxin Testing | Limulus Amebocyte Lysate (LAL) kit (kinetic turbidimetric/chromogenic); Control Standard Endotoxin (CSE); Endotoxin-free consumables. | Ensures parenteral nanoformulations meet USP pyrogenicity safety limits, a critical release criterion. |
| Dispersion Media | Phosphate Buffered Saline (PBS), cell culture media (e.g., DMEM + 10% FBS), biorelevant media (FaSSIF/FeSSIF). | Simulates the biological environment for in vitro characterization, assessing stability and agglomeration state. |
| Reference Materials | NIST Gold Nanoparticle Reference Materials (e.g., RM 8011, 8012, 8013); Latex size standards. | Provides calibrants for instrument verification and method validation, ensuring data accuracy and regulatory compliance. |
Within the complex landscape of nanotechnology product development, strategic regulatory engagement is a critical determinant of success. The U.S. Food and Drug Administration (FDA) offers two key formal consultation programs—Pre-Investigational New Drug (Pre-IND) Meetings and INTERACT (INitial Targeted Engagement for Regulatory Advice on CBER producTs)—to facilitate early, non-binding discussions with sponsors. For nanotechnology-based therapeutics, diagnostics, and combination products, these meetings are invaluable for aligning development plans with regulatory expectations, particularly concerning novel characterization methods, safety assessments, and manufacturing controls unique to nanoscale materials.
Comparative Analysis: INTERACT vs. Pre-IND Meetings
The choice between an INTERACT and a Pre-IND meeting depends on the stage of development and the type of feedback required. The following table summarizes the key quantitative and qualitative parameters of each program.
Table 1: Comparative Summary of FDA INTERACT and Pre-IND Meeting Programs
| Parameter | INTERACT Meeting | Pre-IND Meeting |
|---|---|---|
| Development Stage | Very early (preclinical, pre-IND submission) | Later stage (completed preclinical studies, immediately pre-IND submission) |
| Primary Purpose | Preliminary, informal advice on initial development plans, chemistry, manufacturing, and controls (CMC), and preclinical studies. | Formal, binding advice on specific development plans and the adequacy of data to support an IND submission. |
| Timing (FDA Goal) | Scheduling within 21 calendar days of request. | Written response within 60 days of meeting. |
| Formality | Informal, non-binding advice. No official minutes. | Formal, binding agreement if consensus is reached. Official minutes are generated. |
| Meeting Format | Typically a teleconference. | Can be face-to-face, teleconference, or videoconference. |
| Submission Package | Limited (e.g., 5-10 page summary + key supporting data). | Comprehensive (detailed summary + complete preclinical/CMC data packages). |
| Best Suited For (Nanotech) | Initial feedback on novel platform, early toxicology strategy, or innovative characterization methods. | Final agreement on IND-enabling study design, clinical protocol, and product specifications. |
Application Notes for Nanotechnology Product Development
Key Considerations for Meeting Requests
- Product Characterization: Be prepared to discuss robust physicochemical characterization (size, surface charge, morphology, drug release kinetics) using orthogonal methods. Propose methods early.
- Safety Assessment: Anticipate questions on novel toxicology endpoints, immunogenicity, and biodistribution studies specific to the nanomaterial's pharmacokinetics.
- CMC Strategy: Outline control strategies for critical quality attributes (CQAs) related to nanoscale complexity, such as batch-to-batch consistency, sterility, and stability.
Protocol for Preparing and Executing a Successful Meeting
Protocol 1: Strategic Preparation for an FDA INTERACT/Pre-IND Meeting on a Nanotherapeutic
Objective: To systematically prepare for and conduct an early-engagement meeting with the FDA to obtain actionable feedback on the development plan for a novel liposomal nanoparticle drug product.
Materials & Reagents:
- Comprehensive internal development plan document.
- Collated preclinical data (in vitro efficacy, PK/PD, preliminary toxicology).
- Draft Investigator's Brochure (for Pre-IND).
- Draft product characterization data suite.
Procedure:
- Internal Alignment (Week 1-2): Convene a cross-functional team (Regulatory, CMC, Nonclinical, Clinical) to draft a consensus development plan and identify critical questions for the FDA.
- Question Refinement (Week 3): Draft specific, focused, and non-leading questions. Categorize them by discipline (CMC, Pharmacology/Toxicology, Clinical). Limit to 5-7 primary questions.
- Package Assembly (Week 4-6):
- For INTERACT: Prepare a concise briefing package (≤ 15 pages) including product description, mechanism of action, preliminary data summary, proposed development pathway, and specific questions.
- For Pre-IND: Prepare a comprehensive briefing package (≥ 50 pages) with detailed summaries of chemistry, manufacturing, controls, pharmacology, toxicology, and proposed clinical protocol, plus the complete set of specific questions.
- Formal Request Submission: Submit the meeting request and briefing package via the appropriate FDA portal (e.g., CDER NextGen Portal, CBER Office of Communication, Outreach and Development). Adhere to specified page limits and formatting.
- Pre-Meeting Preparation (Upon FDA Acknowledgement): Conduct internal rehearsals. Designate a primary speaker and note-taker for each discipline. Prepare succinct slide summaries (if applicable).
- Meeting Conduct: Adhere to the agreed agenda. Present background briefly (≤ 15 minutes). Focus the discussion on seeking clarity on FDA's responses to the submitted questions.
- Post-Meeting Follow-up: Distribute internal meeting notes within 24 hours. Formalize the agreed-upon path forward. For Pre-IND, await official FDA meeting minutes, compare with internal notes, and resolve any discrepancies in writing with the FDA.
Visual Workflows and Pathways
Title: Decision and Workflow for FDA Early Engagement Meetings
Title: Key Nanotech Development Domains for FDA Discussion
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for Nanotechnology Characterization & Safety Assessment
| Reagent/Material | Supplier Examples | Primary Function in Nanotech Development |
|---|---|---|
| Dynamic Light Scattering (DLS) & Zeta Potential Standards | Malvern Panalytical, Horiba | Calibrating instruments for accurate nanoparticle size (hydrodynamic diameter) and surface charge (zeta potential) measurement. |
| Chromatography Columns (SEC, AF4) | Waters, Wyatt, Postnova | Separating nanoparticles by size for purity analysis and aggregation assessment (Size Exclusion Chromatography) or detailed sub-population resolution (Asymmetric Flow Field-Flow Fractionation). |
| Reference Nanomaterials (NIST Traceable) | NIST, nanoComposix | Acting as positive controls for characterization assays (e.g., size, shape) and toxicology studies to benchmark behavior and instrument performance. |
| Endotoxin Detection Kits (LAL) | Lonza, Associates of Cape Cod | Quantifying bacterial endotoxin levels, a critical safety test for injectable nanotherapeutics, as per USP <85> guidelines. |
| In Vitro Toxicology Assay Kits (Cell Viability, ROS, Cytokine) | Thermo Fisher, Abcam, R&D Systems | Screening for nanoparticle-induced cytotoxicity, oxidative stress, and immunogenicity (pyrogenicity) in relevant cell models prior to animal studies. |
| Animal Models for Biodistribution/PK Studies | Jackson Laboratory, Charles River | Utilizing immunocompetent or disease-specific models to study nanoparticle pharmacokinetics, targeting, and accumulation in organs. |
| Stable Isotope or Fluorophore Labels for Tracking | Creative Diagnostics, Lumiprobe | Conjugating tags (e.g., DyLight dyes, Zr-89 for PET) to nanoparticles to enable sensitive in vitro and in vivo tracking and quantification. |
Identifying Critical Quality Attributes (CQAs) for Nanomaterials from the Start
Within the paradigm of FDA-industry consultation for nanotechnology product development, the early identification of Critical Quality Attributes (CQAs) is a fundamental regulatory and scientific expectation. CQAs are physical, chemical, biological, or microbiological properties that must be within an appropriate limit, range, or distribution to ensure desired product quality, safety, and efficacy. For nanomedicines, CQAs are intrinsically linked to their complex, multifunctional nature. This Application Note details protocols and experimental workflows to systematically define CQAs at the earliest stages of nanomaterial development, aligning with Quality by Design (QbD) principles advocated by regulatory agencies.
Core Nanomaterial CQAs: Quantitative Targets & Measurement Protocols
The following table summarizes primary CQA categories and target ranges informed by current regulatory guidance and literature.
Table 1: Foundational CQAs for Nanomaterials in Drug Development
| CQA Category | Specific Attribute | Typical Target Range/Value | Analytical Method |
|---|---|---|---|
| Physical | Particle Size & Distribution (Hydrodynamic Diameter) | 10-200 nm (system-dependent); PDI < 0.2 | Dynamic Light Scattering (DLS) |
| Particle Size & Morphology (Primary) | As designed (e.g., spherical, rod) | Transmission Electron Microscopy (TEM) | |
| Surface Charge (Zeta Potential) | ±10 - ±30 mV for colloidal stability | Electrophoretic Light Scattering | |
| Drug Loading Capacity & Efficiency | > 5% w/w; Efficiency > 80% | HPLC/UV-Vis after separation | |
| Chemical | Purity & Composition | > 95% (excipients, ligands) | NMR, Mass Spectrometry |
| Surface Chemistry / Ligand Density | Target: 1-5 ligands/nm² | XPS, NMR, Colorimetric assay | |
| Degradation Products | < 2% related substances | HPLC, SEC | |
| Biological | Sterility & Endotoxin | Sterile; Endotoxin < 0.25 EU/mL | USP <71>, LAL/ recombinant assay |
| In Vitro Potency (e.g., Target Binding) | IC50/EC50 within 2-fold of reference | ELISA, Surface Plasmon Resonance | |
| In Vitro Release Profile | Matches desired kinetics (e.g., sustained) | Dialysis, USP Apparatus 4 |
Experimental Protocols for Key CQA Assessments
Protocol 2.1: Comprehensive Size and Charge Analysis (DLS & ELS) Objective: Determine hydrodynamic diameter (size), polydispersity index (PDI), and zeta potential of nanoparticles in suspension. Materials: Nanoparticle dispersion, appropriate buffer (e.g., 1xPBS, pH 7.4), disposable sizing cuvettes, disposable folded capillary zeta cells, DLS/Zeta potential analyzer. Procedure:
- Sample Preparation: Dilute the nanoparticle sample in a filtered (0.1 µm) appropriate buffer to achieve a count rate within the instrument's optimal sensitivity range. Perform dilution in triplicate.
- Equilibration: Allow the sample and instrument to equilibrate to 25.0 ± 0.1°C for 300 seconds.
- DLS Measurement: Transfer sample to a clean sizing cuvette. Perform a minimum of 12 sub-runs. Record the Z-average diameter (intensity-weighted mean) and the PDI.
- Zeta Potential Measurement: Rinse a folded capillary cell 3x with filtered buffer. Load the same diluted sample. Apply a field strength of ~15-20 V/cm. Perform a minimum of 100 runs. Record the mean zeta potential and electrophoretic mobility.
- Data Analysis: Report the mean ± standard deviation of the triplicate measurements. A PDI > 0.3 indicates a highly polydisperse system requiring further purification.
Protocol 2.2: Determination of Drug Loading by Direct and Indirect Methods Objective: Quantify the amount of active pharmaceutical ingredient (API) encapsulated per unit mass of nanoparticle. Materials: Nanoparticle dispersion, ultracentrifuge, HPLC system with UV/Vis detector, appropriate organic solvents for nanoparticle disruption (e.g., acetonitrile, methanol), 10 kDa molecular weight cut-off (MWCO) centrifugal filters. Procedure A (Direct - After Digestion/Dissolution):
- Disruption: Transfer 200 µL of nanoparticle suspension to a vial. Add 800 µL of organic solvent to fully dissolve/disrupt the nanoparticle matrix. Vortex for 5 minutes.
- Dilution: Dilute the solution appropriately with mobile phase compatible solvent.
- Quantification: Inject onto HPLC system and quantify API against a validated standard curve. Calculate loading capacity (LC) as (mass of API in NPs / total mass of NPs) * 100%.
Procedure B (Indirect - Free Drug Separation):
- Separation: Load 500 µL of nanoparticle suspension into a 10 kDa MWCO centrifugal filter. Centrifuge at 14,000 x g for 30 min.
- Collection: Collect the filtrate containing unencapsulated (free) drug.
- Quantification: Analyze the filtrate by HPLC to determine free drug concentration. Calculate encapsulated drug = total drug - free drug. Calculate encapsulation efficiency (EE) as (mass of encapsulated API / total mass of API fed) * 100%.
Visualization of CQA-Driven Development Workflow
Diagram Title: QbD Workflow for Nanomaterial CQAs (85 chars)
Mapping CQAs to Biological Performance & Signaling Pathways
A key CQA for targeted nanomaterials is ligand density, which directly influences cellular uptake and downstream signaling.
Diagram Title: CQA Impact on Cellular Signaling Pathway (77 chars)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for CQA Characterization
| Item | Function in CQA Assessment |
|---|---|
| NIST Traceable Size Standards (e.g., 60nm, 100nm polystyrene beads) | Calibration and validation of DLS, NTA, and electron microscopy instruments for accurate size measurement. |
| Zeta Potential Transfer Standard (e.g., -50mV ± 5mV latex) | Verification of instrument performance for surface charge measurements. |
| Endotoxin-Free Water & Vials | Preparation of samples for sterility and endotoxin testing to avoid contamination. |
| Size Exclusion Chromatography (SEC) Columns (e.g., Sepharose CL-4B, Superose) | Purification of nanoparticles from unencapsulated drug or free ligands for accurate loading/efficiency assays. |
| Surface Plasmon Resonance (SPR) Chips (e.g., CM5 sensor chip) | Label-free, quantitative analysis of targeting ligand affinity (kon, koff, KD) to the biological target. |
| Fluorescently-Labeled Lipid/Polymer Conjugates | Tracer components for imaging intracellular trafficking and quantifying cellular uptake via flow cytometry. |
| Protease/Enzyme Activity Assay Kits | Assessment of nanoparticle impact on enzyme function or simulation of drug release in biological matrices. |
| Stable Isotope-Labeled Precursors (e.g., 13C-labeled polymers) | Enables precise tracking of nanoparticle metabolism and degradation products via Mass Spectrometry. |
Within the context of FDA-industry consultation for nanotechnology product development, selecting the appropriate regulatory pathway is a critical strategic decision. Nanomedicines, due to their complex physicochemical properties and often novel mechanisms of action, require careful alignment with FDA submission types. The primary pathways are the New Drug Application (NDA), the Biologics License Application (BLA), and the 505(b)(2) application, a specialized type of NDA.
- New Drug Application (NDA) [505(b)(1)]: The full application for a new chemical entity (NCE). For nanomedicines, this is used when the active moiety itself is novel (e.g., a new nanocrystal or a novel nanoparticle-drug conjugate where both components are unapproved). It requires full reports of safety and effectiveness investigations.
- Biologics License Application (BLA): The pathway for biological products, including certain nanomedicines like liposomal vaccines, viral vector nanoparticles for gene therapy, and monoclonal antibodies conjugated to nanoparticles. The determination between NDA and BLA hinges on the "primary mode of action" as defined by the FDA.
- 505(b)(2) Application: A hybrid pathway crucial for many nanomedicine developers. It allows for reliance on the FDA's finding of safety and/or effectiveness for a previously approved ("listed") drug, while submitting new data for the modified product. This is highly relevant for reformulations (e.g., polymeric nanoparticles of an old drug for improved pharmacokinetics), new dosage forms, or new combinations involving nanotechnology.
Table 1: Comparison of Key Regulatory Pathways for Nanomedicines
| Feature | NDA (505(b)(1)) | 505(b)(2) Application | BLA |
|---|---|---|---|
| Legal Basis | FD&C Act, Section 505(b)(1) | FD&C Act, Section 505(b)(2) | PHS Act, Section 351 |
| Appropriate Nanomedicine Example | Novel siRNA-loaded lipid nanoparticle for an unmet need | Paclitaxel albumin-bound nanoparticles (reformulation of an approved chemotherapeutic) | Liposomal-based vaccine or AAV nanoparticle gene therapy |
| Data Requirement | Full, original nonclinical & clinical data | Mixture of original data and literature; can rely on FDA's prior findings on a listed drug | Full data package for the biological product; often includes extensive CMC & immunogenicity data |
| Development Time & Cost | Highest (Typically >10 years, >$1B) | Moderate to High (Reduced vs. NDA due to reliance on existing data) | Very High (Complex manufacturing, stringent characterization) |
| Exclusivity Periods | 5-year New Chemical Entity (NCE); 3-year New Clinical Investigation | 3-year New Clinical Investigation (often applicable) | 12-year Reference Product Exclusivity (for biologics); 4-year Data Exclusivity |
| Primary Regulatory Focus | Novelty, full safety/efficacy profile | Bridging studies demonstrating how the modified product relates to the listed drug | Manufacturing process (consistency), immunogenicity, biological activity |
Experimental Protocols for Critical Characterization Studies
The following protocols are essential for generating data to support any of the aforementioned regulatory submissions, particularly in demonstrating comparability (for 505(b)(2)) or novel characteristics (for NDA/BLA).
Protocol 1: Critical Quality Attribute (CQA) Profiling of Nanomedicine Formulation
Objective: To characterize the physicochemical properties that define the identity, strength, quality, purity, and potency of the nanomedicine product.
Materials: See "The Scientist's Toolkit" (Section 4).
Methodology:
- Sample Preparation: Dilute the nanomedicine formulation in appropriate buffer (e.g., PBS, 5% sucrose) to a target concentration within the dynamic range of each instrument. Perform in triplicate.
- Particle Size & Distribution (by DLS): Equilibrate the instrument at 25°C. Load 1 mL of diluted sample into a disposable cuvette. Perform 3 measurements of 60-second runs each. Record the Z-average hydrodynamic diameter (Z-avg, d.nm) and polydispersity index (PDI).
- Surface Charge (Zeta Potential, by ELS): Load 1 mL of diluted sample into a folded capillary cell. Set the voltage and perform at least 10-20 runs. Record the average zeta potential (ζ, mV) and conductivity.
- Particle Concentration & Morphology (by NTA): Syringe-inject the diluted sample into the sample chamber until the laser path is visible. Capture a 60-second video under constant flow conditions. Analyze at least 500 tracks to determine particle concentration (particles/mL) and generate a size distribution profile.
- Drug Loading & Entrapment Efficiency (by HPLC/UV-Vis):
- Total Drug: Dilute and lyse an aliquot of the formulation with an organic solvent (e.g., acetonitrile/methanol). Vortex and centrifuge. Analyze the supernatant against a standard curve.
- Free (Unentrapped) Drug: Separate free drug via size-exclusion chromatography (e.g., minicolumn centrifugation) or ultrafiltration. Analyze the filtrate.
- Calculate: Drug Loading (%) = (Mass of entrapped drug / Mass of total lipids+polymer+drug) x 100. Entrapment Efficiency (%) = (Mass of entrapped drug / Total mass of drug added) x 100.
Protocol 2: In Vitro Drug Release Kinetics Using Dialysis
Objective: To establish a correlation between nanoparticle characteristics and drug release profile, a key element for demonstrating controlled release in a 505(b)(2) application or defining a novel product.
Methodology:
- Setup: Place a measured volume (e.g., 1 mL) of nanomedicine formulation into a pre-soaked dialysis membrane tube (MWCO selected based on free drug size, typically 12-14 kDa).
- Release Medium: Immerse the sealed tube in a large volume (e.g., 200 mL, ensuring sink conditions) of release medium (e.g., PBS at pH 7.4, or PBS with 0.1% w/v Tween 80) in a shaking water bath maintained at 37°C ± 0.5°C.
- Sampling: At predetermined time intervals (e.g., 0.5, 1, 2, 4, 8, 12, 24, 48, 72 h), withdraw 1 mL aliquot from the external release medium and replace with an equal volume of fresh, pre-warmed medium.
- Analysis: Quantify the amount of drug released in each aliquot using a validated HPLC or UV-Vis method. Plot cumulative drug release (%) versus time to generate the release profile. Fit data to kinetic models (e.g., zero-order, first-order, Higuchi).
Regulatory Decision & Development Pathways
Diagram Title: Nanomedicine Regulatory Pathway Decision Tree
Diagram Title: Integrated Nanomedicine Development Timeline
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Nanomedicine Characterization
| Item / Reagent | Function & Relevance to Regulatory Filing |
|---|---|
| Dynamic Light Scattering (DLS) Instrument | Measures hydrodynamic diameter (size) and polydispersity index (PDI). Critical CQA for stability and batch-to-batch consistency. |
| Zeta Potential Analyzer | Measures surface charge (zeta potential). Predicts colloidal stability and indicates surface modification. Key CQA. |
| Nanoparticle Tracking Analysis (NTA) System | Provides particle concentration and high-resolution size distribution based on light scattering and Brownian motion. Essential for low-concentration samples (e.g., gene therapies). |
| Size-Exclusion Chromatography (SEC) Columns | For purification and separation of free drug/ligand from nanoparticles. Used in entrapment efficiency and drug loading assays. |
| Dialysis Membranes (Various MWCO) | Used for in vitro drug release studies. Data supports controlled-release or modified PK claims in applications. |
| Phospholipid & Polymer Standards | High-purity lipids (DSPC, DOPE, Cholesterol) and polymers (PLGA, PEG) for formulation. GMP-grade required for clinical material. |
| Stability Chambers | For ICH guideline stability testing (e.g., 25°C/60%RH, 5°C). Long-term and accelerated stability data is mandatory for all applications. |
| Reference Listed Drug (RLD) Substance | For 505(b)(2) applications, the approved drug product is used as a comparator in bioanalytical and in vitro equivalence studies. |
Within the context of advancing nanotechnology product development for FDA-regulated therapies, rigorous Chemistry, Manufacturing, and Controls (CMC) principles form the non-clinical foundation for ensuring product quality, safety, and efficacy. For nanomedicines (e.g., liposomes, polymeric nanoparticles, inorganic nanoparticles), CMC challenges are magnified due to complex physicochemical attributes and biological interactions. This document outlines critical CMC considerations, supported by application notes and experimental protocols, tailored for researchers and drug development professionals navigating the intersection of nanotechnology and regulatory science.
Key CMC Challenges for Nanotechnology Products
Nanoparticle therapeutics exhibit complex structure-activity relationships. Critical quality attributes (CQAs) must be identified and controlled throughout development.
Table 1: Primary CMC Challenges and Associated CQAs for Nanotechnology-Based Drug Products
| CMC Challenge | Critical Quality Attribute (CQA) | Typical Target Specification | Impact on Safety/Efficacy |
|---|---|---|---|
| Particle Size & Distribution | Mean particle diameter, Polydispersity Index (PDI) | e.g., 100 nm ± 10 nm, PDI < 0.2 | Biodistribution, targeting, clearance rate |
| Surface Characteristics | Zeta potential, PEG density, ligand conjugation efficiency | e.g., Zeta: -10 to -30 mV | Stability, protein corona formation, cellular uptake |
| Drug Loading & Release | Drug payload (w/w%), encapsulation efficiency, in vitro release profile | e.g., > 90% encapsulation, sustained release over 24h | Therapeutic dose, pharmacokinetics, efficacy |
| Structural Integrity & Morphology | Particle morphology (TEM/SEM), lamellarity (liposomes), crystallinity | Spherical, uniform, defined internal structure | Drug retention, stability, manufacturability |
| Purity & Impurities | Residual solvents, metal catalysts (inorganics), endotoxin levels | Per ICH Q3 guidelines, endotoxin < 0.5 EU/mL | Safety, immunogenicity |
Application Note: Characterization of Nanoparticle Size and Surface Charge
Objective: To reliably determine the mean particle size, size distribution (PDI), and zeta potential of a nanoparticle formulation as key CQAs for regulatory filing.
Protocol 1: Dynamic Light Scattering (DLS) for Size and PDI
- Principle: Measures Brownian motion to calculate hydrodynamic diameter.
- Materials: Nanoparticle dispersion, appropriate buffer (e.g., 1xPBS, pH 7.4), disposable sizing cuvettes, DLS instrument (e.g., Malvern Zetasizer).
- Procedure:
- Dilution: Dilute nanoparticle sample in filtered (0.1 µm) buffer to achieve an optimal scattering intensity. Avoid multiple scattering.
- Equilibration: Allow sample and instrument to equilibrate to 25°C for 300 seconds.
- Measurement: Transfer to cuvette, place in instrument. Set parameters: material RI: 1.59, dispersant RI: 1.33, viscosity: 0.8872 cP.
- Run: Perform a minimum of 3 runs per sample, each consisting of 10-15 sub-runs.
- Analysis: Record the Z-average diameter (intensity-weighted mean) and the Polydispersity Index (PDI). Report as Mean ± S.D. (n≥3 independent batches).
Protocol 2: Phase Analysis Light Scattering (PALS) for Zeta Potential
- Principle: Measures electrophoretic mobility in an applied field to calculate zeta potential.
- Materials: Nanoparticle dispersion, clear disposable zeta cell, zeta potential instrument.
- Procedure:
- Sample Preparation: Use the same diluted sample as for DLS.
- Loading: Inject sample into a clean, dry zeta cell using a syringe, ensuring no air bubbles.
- Measurement: Set parameters: Smoluchowski model, F(Ka) = 1.5. Perform measurements at a fixed stationing position.
- Run: Conduct a minimum of 3 runs with >12 sub-runs each. The instrument automatically calculates zeta potential (mV).
- Analysis: Report the mean zeta potential and its standard deviation. The magnitude indicates colloidal stability (>|30| mV: high stability).
Application Note: Assessing Drug Loading andIn VitroDrug Release
Objective: To quantify the amount of active pharmaceutical ingredient (API) associated with nanoparticles and characterize its release kinetics under physiologically relevant conditions.
Protocol 3: Determination of Encapsulation Efficiency and Drug Loading
- Principle: Separate unencapsulated/free drug from nanoparticle-associated drug and quantify both fractions.
- Materials: Ultracentrifuge with appropriate rotors, centrifugal filter devices (MWCO 10-50 kDa), HPLC system with UV/Vis detector, mobile phase solvents.
- Procedure:
- Separation of Free Drug: Aliquot 500 µL of nanoparticle suspension. Ultracentrifuge at 150,000 x g for 45 min at 4°C. Carefully collect the supernatant (contains free drug). Alternatively, use centrifugal filtration.
- Lysis of Nanoparticles: Resuspend the pellet in 500 µL of a lysing agent (e.g., 1% Triton X-100 in buffer or 70% ethanol/30% water). Vortex vigorously to ensure complete disruption.
- Drug Quantification: Analyze both the supernatant (free drug) and the lysate (encapsulated drug) using a validated HPLC-UV method.
- Calculation:
- Encapsulation Efficiency (%) = (Mass of encapsulated drug / Total mass of drug input) x 100
- Drug Loading (w/w%) = (Mass of encapsulated drug / Total mass of nanoparticles) x 100
- Data Presentation: Results should be tabulated for multiple production batches.
Protocol 4: In Vitro Drug Release Using Dialysis
- Principle: Uses a dialysis membrane to contain nanoparticles while allowing released drug to diffuse into a sink medium.
- Materials: Dialysis tubing (appropriate MWCO, e.g., 12-14 kDa), release medium (e.g., PBS with 0.5% w/v Tween 80 to maintain sink conditions), shaking water bath.
- Procedure:
- Setup: Load 1 mL of nanoparticle suspension into pre-hydrated dialysis tubing. Seal ends tightly.
- Incubation: Immerse the dialysis bag in 200 mL of pre-warmed release medium (37°C) with gentle agitation (50 rpm).
- Sampling: At predetermined time points (e.g., 0.5, 1, 2, 4, 8, 24, 48 h), withdraw 1 mL aliquots from the external medium and replace with fresh pre-warmed medium.
- Analysis: Quantify drug concentration in each aliquot using HPLC.
- Data Analysis: Calculate cumulative drug release (%) over time. Plot release profile. Fit data to appropriate kinetic models (e.g., zero-order, first-order, Higuchi, Korsmeyer-Peppas) to understand release mechanism.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Nanomedicine CMC Characterization
| Item | Function in CMC Studies | Example/Notes |
|---|---|---|
| Phospholipids (e.g., DSPC, DPPC) | Primary bilayer component for liposomal NPs. Dictates membrane rigidity, phase transition temperature. | High-purity (>99%) sources required for batch consistency. |
| Polymeric Excipients (e.g., PLGA, PEG-PLGA) | Biodegradable polymer core for drug encapsulation and controlled release. | Varying molecular weights and LA:GA ratios affect degradation rate. |
| PEGylated Lipids (e.g., DSPE-PEG2000) | Provides steric stabilization ("stealth" effect) to reduce macrophage uptake and prolong circulation. | Critical for controlling protein corona formation. |
| Functional Ligands (e.g., Folate, RGD peptide) | Enables active targeting to specific cell receptors. Must be conjugated with high reproducibility. | Conjugation chemistry (e.g., maleimide-thiol) must be validated and controlled. |
| Size Exclusion Chromatography (SEC) Columns | Purify nanoparticles from unencapsulated drugs, free ligands, or aggregates. | Sepharose CL-4B or Sephacryl S-1000 are common for large nanoparticles. |
| DLS/Zeta Potential Standards | Validate instrument performance (size and zeta) prior to sample analysis. | Polystyrene latex beads (e.g., 100 nm) and zeta potential transfer standard. |
| Stability Study Buffers | Assess physical and chemical stability of NPs under ICH conditions (e.g., pH, ionic strength). | PBS, histidine-sucrose, or other formulation-specific buffers. |
Regulatory Pathway Diagram
Title: Regulatory CMC Pathway for Nanomedicine Development
Nanoparticle Characterization Workflow
Title: Integrated CMC Characterization Workflow for Nanoparticles
Practical Strategies for Developing and Characterizing Nano-Formulations
Essential Analytical Methods for Physicochemical Characterization (Size, Charge, Stability)
This document provides detailed application notes and protocols for the essential analytical methods required for the physicochemical characterization of nanotechnology-based products. Within the context of FDA industry consultation and regulatory guidelines (e.g., FDA Guidance for Industry: Drug Products, Including Biological Products, that Contain Nanomaterials, 2022), these methods are critical for demonstrating critical quality attributes (CQAs) that impact safety, efficacy, and stability.
Dynamic Light Scattering (DLS) for Hydrodynamic Size Distribution
Application Note: DLS is the primary technique for determining the hydrodynamic diameter (Z-average) and size distribution (polydispersity index, PDI) of nanoparticles in suspension. It is essential for batch-to-batch consistency and early detection of aggregation.
Protocol: Sample Preparation and Measurement
- Sample Dilution: Dilute the nanoformulation in the appropriate buffer (e.g., PBS, 1 mM KCl) to achieve an optimal scattering intensity. The final concentration should yield a count rate between 200-500 kcps for most instruments.
- Filtration: Filter the diluent through a 0.1 or 0.2 µm membrane filter to remove dust.
- Equilibration: Allow the sample and measurement cell (typically a disposable cuvette) to equilibrate to the instrument temperature (typically 25°C) for 2 minutes.
- Measurement: Perform a minimum of 3-10 measurements per sample, with each run lasting 10-60 seconds. Include an angle measurement (commonly 173° backscatter) to minimize multiple scattering.
- Data Analysis: Report the Z-average diameter (intensity-weighted mean), the PDI, and the intensity size distribution plot. The PDI threshold for a monodisperse sample is generally <0.2.
Table 1: Representative DLS Data for a Liposomal Formulation
| Batch ID | Z-Average Diameter (nm) | PDI | Result Interpretation |
|---|---|---|---|
| Lipo-001 | 102.4 ± 1.2 | 0.08 ± 0.01 | Monodisperse, acceptable. |
| Lipo-002 | 156.7 ± 15.8 | 0.32 ± 0.05 | Polydisperse, indicates aggregation. |
| Lipo-003 | 99.8 ± 0.9 | 0.06 ± 0.01 | Monodisperse, acceptable. |
Diagram: DLS Experimental Workflow
Title: DLS Measurement and Data Analysis Process
Laser Doppler Microelectrophoresis for Zeta Potential
Application Note: Zeta potential indicates the surface charge of nanoparticles, predicting colloidal stability. A magnitude greater than |±30| mV typically indicates good electrostatic stability.
Protocol: Measurement via Phase Analysis Light Scattering (M3-PALS)
- Cell Selection & Loading: Use a clean, dedicated zeta potential folded capillary cell. Inject 0.8-1.0 mL of the undiluted or minimally diluted sample using a syringe, avoiding air bubbles.
- Instrument Setup: Select the appropriate material model (e.g., lipid, polymer) and solvent parameters (viscosity, dielectric constant). Set temperature to 25°C.
- Voltage Application: Apply a field strength of ~10-20 V/cm. The instrument will use PALS to measure particle velocity.
- Measurement & Calculation: Perform a minimum of 10-30 runs per sample. The software uses the Henry equation to calculate zeta potential from electrophoretic mobility. Report the mean zeta potential and its standard deviation.
Table 2: Zeta Potential Stability Study Under Stress Conditions
| Storage Condition (4 Weeks) | Initial ZP (mV) | Final ZP (mV) | Δ ZP | Stability Indication |
|---|---|---|---|---|
| 4°C, pH 7.4 | -42.5 ± 2.1 | -41.8 ± 3.0 | -0.7 | Stable |
| 25°C, pH 7.4 | -43.0 ± 1.8 | -35.2 ± 5.1 | -7.8 | Moderately Stable |
| 25°C, pH 5.5 | -42.1 ± 2.3 | -15.6 ± 8.4 | -26.5 | Unstable (Aggregation) |
Diagram: Factors Influencing Zeta Potential & Stability
Title: Zeta Potential Determinants and Stability Outcomes
Nanoparticle Tracking Analysis (NTA) for Concentration and Size
Application Note: NTA provides direct visualization and analysis of nanoparticles in liquid, yielding particle-by-particle size and an estimate of concentration (particles/mL), crucial for dosing.
Protocol: Sample Analysis via NTA
- Critical Dilution: Dilute sample in particle-free buffer to achieve 20-100 particles per frame. This often requires a dilution factor of 10,000 to 1,000,000x.
- Syringe Loading: Use a sterile syringe to inject 0.3-1.0 mL of diluted sample into the sample chamber. Ensure no air bubbles are introduced.
- Camera Setup: Adjust the camera level and detection threshold to clearly visualize individual particle scattering centers against the background.
- Video Capture: Record three 60-second videos of different sample portions. Maintain constant temperature.
- Data Processing: Use software to track the Brownian motion of each particle. The Stokes-Einstein equation is applied to calculate the diameter. Report the mode, mean, D10, D50 (median), D90, and concentration.
Table 3: NTA vs. DLS Comparison for a Polymeric Nanoparticle Sample
| Parameter | NTA Result | DLS Result | Note |
|---|---|---|---|
| Primary Size (Mode) | 78.2 nm | N/A | Most frequent size by count. |
| Mean Size | 85.4 ± 12.3 nm | 96.7 ± 3.1 nm (Z-Ave) | NTA is number-weighted; DLS is intensity-weighted. |
| Concentration | (3.2 ± 0.4) x 10^11 part./mL | N/A | Critical for PK/PD studies. |
| Sensitivity to Aggregates | High (visualized) | Very High (dominates signal) | NTA can resolve sub-populations. |
Stability Indicating Methods: Accelerated and Real-Time Studies
Protocol: Forced Degradation and Stability Study Design
- Stress Conditions: Aliquot the nanoformulation and expose it to:
- Thermal: 4°C, 25°C, 40°C, and 60°C.
- Photostability: Expose to UV (320-400 nm) and visible light per ICH Q1B.
- Mechanical Stress: Agitation (e.g., 200 rpm) or freeze-thaw cycles (-20°C to 25°C).
- pH Variation: Dilute in buffers of physiologically relevant pH (e.g., 5.0, 7.4).
- Sampling Time Points: T = 0, 1, 2, 4 weeks, and 3, 6 months for real-time.
- Analysis: At each time point, analyze samples using:
- Primary Methods: DLS (for size/PDI) and Zeta Potential.
- Orthogonal Methods: NTA, Tunable Resistive Pulse Sensing (TRPS), or UV-Vis spectroscopy for absorbance shifts.
- Visual Inspection: For precipitation or color change.
- Acceptance Criteria: Define stability thresholds (e.g., Δ Z-Ave < 10%, PDI < 0.25, Δ ZP < 5 mV).
The Scientist's Toolkit: Key Reagent Solutions for Characterization
| Item/Reagent | Function in Characterization |
|---|---|
| Phosphate Buffered Saline (PBS), 1x, Filtered (0.1 µm) | Standard isotonic diluent for size and zeta potential measurements. |
| 1 mM Potassium Chloride (KCl) Solution | Low ionic strength medium for accurate zeta potential measurement. |
| NIST Traceable Size Standards (e.g., 100 nm Polystyrene) | For daily verification and calibration of DLS/NTA instruments. |
| Zeta Potential Transfer Standard (e.g., ±50 mV) | For performance qualification of zeta potential analyzers. |
| Sterile, Particle-Free Water | For dilutions and final rinsing of all vessels and cells. |
| Disposable Zeta Cell & Cuvettes | Eliminates cross-contamination and ensures measurement consistency. |
| Syringe Filters (0.1 µm PES membrane) | For critical filtration of buffers to remove particulate interference. |
Application Notes
This document details critical protocols for scaling nanotechnology-based drug products (NDPs) from laboratory to commercial manufacturing, with an emphasis on contamination control, as per FDA guidance and industry best practices for pre-market consultation.
1. Critical Quality Attributes (CQAs) for Nanotechnology Scale-Up Successful tech transfer requires identifying and monitoring CQAs that impact safety and efficacy. For NDPs, these often include particle size distribution, zeta potential, drug loading, and sterility/endotoxin levels.
2. Contamination Control Strategy (CCS) Framework A holistic CCS is mandated for aseptic processes. It encompasses design of facilities and equipment, environmental monitoring, vessel and component preparation, and personnel training, with a focus on controlling particulate (including nanomaterial) and microbiological contamination.
Table 1: Key Process Parameters & Target Ranges for Lipid Nanoparticle (LNP) Scale-Up
| Process Parameter | Lab Scale (10 mL) | Pilot Scale (10 L) | Commercial Scale (100 L) | Control Strategy |
|---|---|---|---|---|
| Mixing Flow Rate (T-Junction) | 10 mL/min | 5 L/min | 50 L/min | In-line PAT monitoring |
| Total Mixing Time | 2 min | 5 min | 8 min | Fixed parameter |
| Temperature | 25°C ± 2°C | 25°C ± 1°C | 25°C ± 0.5°C | Jacketed vessel control |
| Final Particle Size (Z-Avg) | 80-100 nm | 85-105 nm | 90-100 nm | Real-time DLS/SLS |
| PDI (Polydispersity) | ≤ 0.15 | ≤ 0.18 | ≤ 0.15 | Acceptance criterion |
3. Environmental Monitoring (EM) Data Analysis Routine EM provides trend data for contamination control. Action limits are defined per ISO 14644 and EU GMP Annex 1.
Table 2: Example Environmental Monitoring Action Limits for Grade A/B Areas
| Location | Viable Air (CFU/m³) | Non-Viable Particles (≥0.5 μm/m³) | Surface Viable (CFU/contact plate) | Settle Plates (CFU/4 hours) |
|---|---|---|---|---|
| Grade A (At Rest) | <1 | 3,520 | 1 | <1 |
| Grade A (In Operation) | <1 | 3,520 | 1 | <1 |
| Grade B (At Rest) | 10 | 3,520 | 5 | 5 |
| Grade B (In Operation) | 10 | 352,000 | 5 | 5 |
Experimental Protocols
Protocol 1: Scale-Down Model for Sterility Assurance Validation
Objective: To validate the efficacy of the aseptic filling process using a microbiological challenge (media fill). Materials: Sterile growth medium (e.g., TSB), production-line filling equipment (scale-down model), environmental monitoring equipment, incubators. Procedure:
- Setup: Conduct the media fill under conditions that fully simulate the routine aseptic manufacturing process, including duration, number of personnel, and interventions.
- Execution: Aseptically fill sterile growth medium into the final product containers using the standard process. Perform all planned interventions.
- Controls: Include positive controls (inoculated medium) and negative controls (unopened medium containers).
- Incubation: Incubate all filled containers at 20-25°C for 7 days, then at 30-35°C for 7 days.
- Inspection: Visually inspect for microbial growth (turbidity). Any contaminated unit is considered a failure. The batch passes if contamination rate is <0.1% with 95% confidence (based on batch size).
- Documentation: Record all EM data during the process and correlate with any contaminated units.
Protocol 2: Determination of Sub-Visible Particulate Matter in NDPs
Objective: Quantify sub-visible particles (2-10 μm) per USP <788> and <789> to assess contamination from process or packaging. Materials: Light obscuration particle count tester (e.g., HIAC), syringe assembly, particle-free water, magnetic stirrer. Procedure:
- Calibration: Calibrate the particle counter using standard size latex spheres.
- Sample Preparation: Gently invert the NDP final container 20 times. For viscous products, dilute with particle-free fluid as validated.
- Analysis: Place sample on a stirrer. Draw a minimum of 5 mL through the sensor at a rate of 10-20 mL/min. Perform analysis in quadruplicate, discarding the first mL as a rinse.
- Calculation: Report the mean cumulative particle count per container ≥10 μm and ≥25 μm. Acceptance criteria: NDPs for parenteral use must meet limits of ≤6000/container (≥10 μm) and ≤600/container (≥25 μm).
- Out-of-Specification (OOS): An OOS result triggers an investigation per CFR 211.192, assessing equipment, process, and operator factors.
Diagrams
Title: Contamination Control Strategy Flow
Title: LNP Scale-Up Unit Operations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Nanomedicine Process Development & Control
| Item | Function & Application |
|---|---|
| Size Exclusion Chromatography (SEC) Columns (e.g., Superose 6 Increase) | High-resolution separation of nanocarriers from unencapsulated drug/impurities; critical for determining drug loading and aggregation state. |
| Polycarbonate Membrane Filters (e.g., 100 nm, 50 nm) | For extruding liposomes/LNPs to achieve narrow, defined particle size distributions during small-scale development. |
| Tangential Flow Filtration (TFF) Cassettes (e.g., 300 kDa MWCO) | Scalable method for buffer exchange, concentration, and diafiltration of nanoparticle dispersions; used from pilot to commercial scale. |
| Standard Reference Materials (SRMs) for Particle Sizing (NIST Traceable) | Gold or latex nanoparticles of certified size for daily calibration of Dynamic Light Scattering (DLS) and Nanoparticle Tracking Analysis (NTA) instruments. |
| Limulus Amebocyte Lysate (LAL) Reagents (Gel-Clot, Chromogenic, Turbidimetric) | Gold-standard for quantifying bacterial endotoxin levels in raw materials, in-process samples, and final NDP product to ensure pyrogen-free status. |
| Particle-Free Water & Buffers | Essential for background control in sub-visible particle counting (HIAC) and aseptic process simulation (media fills) to avoid false positives. |
| Single-Use Bioprocess Containers & Assemblies | Pre-sterilized bags, tubing, and connectors that minimize cross-contamination, cleaning validation burden, and preparation time during scale-up. |
| Rapid Microbiological Methods (RMM) Kits (e.g., ATP bioluminescence, PCR-based) | For faster detection and identification of microbial contaminants in environmental and process samples compared to traditional culture methods. |
Documentation Best Practices for Regulatory Dossiers (IND, NDA)
This document outlines best practices for compiling Investigational New Drug (IND) and New Drug Application (NDA) dossiers, with a specific focus on the unique challenges presented by nanotechnology-based drug products. Within the context of accelerating nanomedicine development, precise and compliant documentation is not merely administrative but a critical scientific and regulatory function. These application notes provide structured guidance and protocols to ensure data integrity, clarity, and regulatory acceptance, supporting the broader thesis that robust documentation is foundational to successful FDA-industry consultation for novel nanotherapeutics.
Core Documentation Principles for Nanotechnology Dossiers
The CTD/eCTD Framework
The Common Technical Document (CTD) and its electronic version (eCTD) provide the mandatory organizational structure for regulatory submissions. For nanotech products, particular emphasis must be placed on specific modules where characterization and manufacturing data are critical.
Table 1: CTD Module Emphasis for Nanotechnology Products
| CTD Module | Key Nanotech-Specific Documentation Focus | Critical Data Elements |
|---|---|---|
| Module 2: Summaries | Quality Overall Summary (QOS), Nonclinical & Clinical Summaries. Clearly link nanoparticle properties to safety/efficacy. | Physicochemical characterization tables; PK/PD correlations; summary of critical quality attributes (CQAs). |
| Module 3: Quality | Most Critical Section. Detailed information on Drug Substance and Drug Product manufacturing, characterization, and controls. | Comprehensive CQA list; batch analysis data; stability data under relevant conditions; impurity profiles. |
| Module 4: Nonclinical | Toxicology and pharmacokinetics studies must address nanoparticle-specific behavior (e.g., opsonization, RES uptake, novel toxicity). | Biodistribution data (tables by organ); hematology and clinical chemistry results; histopathology findings. |
| Module 5: Clinical | Clinical study reports must detail handling and administration procedures specific to nano-formulations. | Pharmacokinetic parameters (AUC, Cmax, t1/2); immunogenicity data; administration protocol details. |
Document Quality and Data Integrity
- Attributable, Legible, Contemporaneous, Original, and Accurate (ALCOA+): All source data must comply with these principles. For electronic systems, ensure audit trails are enabled and retained.
- Version Control: Implement a strict document management system with clear versioning, effective dates, and change histories.
- Terminology Consistency: Define and consistently use standardized terms for nanoparticle components (e.g., core, coating, ligand, conjugate) throughout the dossier.
Detailed Application Notes: Characterizing a Nanotechnology Drug Product (Module 3 Focus)
Application Note AN-101: Comprehensive Physicochemical Characterization Protocol
Purpose: To systematically characterize the Critical Quality Attributes (CQAs) of a liposomal nanoparticle drug product, establishing a quality target product profile (QTPP).
Table 2: Essential Characterization Parameters & Methods
| CQA Category | Specific Parameter | Recommended Analytical Method | Acceptance Criteria Rationale |
|---|---|---|---|
| Identity & Structure | Particle Morphology | Transmission Electron Microscopy (TEM) | Confirms expected core-shell structure. |
| Chemical Composition | NMR, Mass Spectrometry | Verifies lipid ratios and API-loading. | |
| Size & Distribution | Mean Hydrodynamic Diameter | Dynamic Light Scattering (DLS) | Impacts biodistribution and clearance. |
| Particle Size Distribution (PDI) | DLS | PDI < 0.2 indicates monodisperse population. | |
| Surface Properties | Zeta Potential | Electrophoretic Light Scattering | Predicts colloidal stability and protein binding. |
| Surface Ligand Density | HPLC or Colorimetric Assay | Critical for active targeting efficiency. | |
| Drug Substance | Encapsulation Efficiency (%) | Size Exclusion Chromatography / Ultrafiltration | Directly impacts potency and toxicity. |
| Drug Release Kinetics | In vitro dialysis under physiologic conditions | Predicts in vivo release profile. | |
| Purity & Stability | Aggregation/Precipitation | Visual Inspection, DLS, SEC-MALS | Ensures product stability and safety. |
| Residual Solvents | GC-MS | Must meet ICH Q3C guidelines. |
Experimental Protocol EP-101: Determining Encapsulation Efficiency and Drug Loading
1. Objective: To accurately quantify the percentage of active pharmaceutical ingredient (API) encapsulated within nanoparticles versus free/unencapsulated API. 2. Materials: * Purified nanoparticle suspension. * Appropriate buffer (e.g., PBS, pH 7.4). * Size-exclusion columns (e.g., Sephadex G-50) or centrifugal ultrafiltration devices (MWCO appropriate for nanoparticle retention). * HPLC system with validated method for API quantification. * Centrifuge. 3. Procedure: 1. Total API Measurement: Dilute an aliquot of the nanoparticle suspension with a suitable solvent (e.g., 1:9 dilution in 90% isopropanol/10% water) to disrupt the particles. Vortex vigorously for 5 minutes. Analyze by HPLC to determine total API concentration (Ctotal). 2. Separation of Free API: Using a separate aliquot, apply the nanoparticle suspension to a size-exclusion column pre-equilibrated with buffer. Elute with buffer and collect the nanoparticle fraction (first turbid eluent). Alternatively, use a centrifugal ultrafiltration device: centrifuge per manufacturer instructions; the retentate contains nanoparticles, the filtrate contains free API. 3. Encapsulated API Measurement: Disrupt the nanoparticle fraction from Step 2.2 using the solvent method from Step 2.1. Analyze by HPLC to determine encapsulated API concentration (Cencapsulated). 4. Calculation: * Encapsulation Efficiency (%) = (Cencapsulated / Ctotal) x 100. * Drug Loading (wt%) = (Mass of encapsulated API / Total mass of nanoparticles) x 100. 4. Documentation: Record raw chromatogram data, calculations, and note any deviations. Include method validation parameters (linearity, recovery) in the dossier appendix.
Visualizing Key Concepts & Workflows
Workflow: CQA Identification to Dossier Submission
Key PK Pathways for Nanoparticles
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Materials for Nanoparticle Characterization
| Item | Supplier Examples | Function in Documentation Context |
|---|---|---|
| Standardized Phospholipids | Avanti Polar Lipids, CordenPharma | Ensure batch-to-batch consistency in liposomal formulations; critical for manufacturing reproducibility. |
| PEGylated Lipids (DSPE-PEG) | NOF America, Lipoid GmbH | Provide steric stabilization; particle size and zeta potential are key CQAs dependent on PEG density/chain length. |
| Size Exclusion Chromatography (SEC) Columns | Cytiva (Sephadex), Tosoh Bioscience | Purify nanoparticles from free API or unincorporated materials; essential for measuring encapsulation efficiency. |
| Zeta Potential & Size Standards | Malvern Panalytical, Thermo Fisher | Calibrate and qualify DLS and electrophoretic light scattering instruments; required for method validation. |
| Stable Cell Lines for Targeting Assays | ATCC, academic repositories | Validate active targeting in vitro; documentation must include cell line authentication and passage number. |
| Reference Nanomaterials | National Institute of Standards and Technology (NIST) | Used as comparators for size, shape, and surface charge measurements; strengthens method robustness. |
This application note details the implementation of Quality by Design (QbD) principles, as outlined in ICH Q8(R2), Q9, and Q10, to the development of a liposomal Doxorubicin formulation, analogous to Doxil. The work is contextualized within a thesis on enhancing regulatory success for complex nanotechnology products through proactive FDA consultation and systematic development.
The QbD paradigm shifts quality from a product of testing to an outcome built into the development process. For liposomes, this involves defining a Quality Target Product Profile (QTPP) and identifying Critical Quality Attributes (CQAs) that impact safety and efficacy. These CQAs are then linked to Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) through a systematic risk assessment and experimental design (DoE).
Defining the QTPP and CQAs
The primary QTPP is a sterile liposomal suspension for intravenous administration, designed for extended circulation and targeted delivery to tumor tissues.
Table 1: QTPP and Derived CQAs for a Liposomal Doxorubicin Formulation
| QTPP Element | Target | Associated CQAs |
|---|---|---|
| Dosage Form | Sterile, particulate-free suspension | Appearance, Sub-visible particles, Sterility |
| Route of Administration | Intravenous infusion | Osmolality, pH, Endotoxin levels |
| Pharmacokinetics | Extended circulation, reduced free drug | Drug release rate in vitro, Size (Z-Avg), Polydispersity Index (PDI) |
| Efficacy | High tumor drug accumulation | Total Drug Content, Encapsulation Efficiency, Lipid Composition |
| Safety (Reduced cardiotoxicity) | Low free drug in plasma | Free Drug Concentration, Phospholipid Degradation Products |
Risk Assessment and Initial Screening
A Fishbone (Ishikawa) diagram and a Risk Assessment Matrix were used to identify potential CMAs and CPPs affecting the CQAs. High-risk factors were selected for DoE studies.
Diagram 1: Risk Assessment for Liposome CQAs
Design of Experiments (DoE) and Data Analysis
A two-stage DoE was employed. First, a Fractional Factorial screening design identified significant factors. Second, a Central Composite Design (CCD) optimized these factors.
Primary DoE Objective: To understand the impact of CPPs on the CQAs of vesicle size (Z-Avg, PDI) and encapsulation efficiency (EE%).
Table 2: DoE Factors and Levels for Liposome Process Optimization
| Factor (CPP) | Low Level (-1) | High Level (+1) | Units |
|---|---|---|---|
| A: Hydration Temperature | 50 | 65 | °C |
| B: Hydration Time | 30 | 90 | min |
| C: Extrusion Cycles | 5 | 15 | passes |
| D: Extrusion Pressure | 100 | 500 | psi |
Table 3: Representative DoE Results and Model Coefficients for Key Responses
| Run | A | B | C | D | Z-Avg (nm) | PDI | EE% |
|---|---|---|---|---|---|---|---|
| 1 | -1 | -1 | -1 | -1 | 135 | 0.25 | 78 |
| 2 | +1 | +1 | +1 | +1 | 88 | 0.08 | 95 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| Coefficient (Z-Avg) | Estimate | p-value | Coefficient (EE%) | Estimate | p-value | ||
| Intercept | 92.5 | <0.001 | Intercept | 92.1 | <0.001 | ||
| A (Temp) | -12.3 | 0.003 | C (Cycles) | -8.2 | 0.010 | ||
| C (Cycles) | -18.7 | <0.001 | D (Pressure) | +5.5 | 0.032 | ||
| D (Pressure) | -9.5 | 0.005 | C*D | -3.1 | 0.045 |
Protocol 1: Preparation and Characterization of Liposomes by Thin-Film Hydration and Extrusion
- Materials: See Scientist's Toolkit.
- Lipid Film Formation: Dissolve DSPC, Cholesterol, and PEG-DSPE (molar ratio 55:40:5) in chloroform in a round-bottom flask. Remove solvent via rotary evaporation (40°C) under reduced pressure for 1 hour to form a thin, dry lipid film. Further dry under high vacuum overnight.
- Hydration: Hydrate the film with ammonium sulfate buffer (250 mM, pH 5.5) pre-heated to the target DoE temperature (e.g., 50-65°C). Gently agitate for the specified DoE time (30-90 min) to form a multilamellar vesicle (MLV) dispersion.
- Size Reduction: Subject the MLV dispersion to sequential extrusion through polycarbonate membranes using a thermobarrel extruder. Perform the specified number of DoE cycles (e.g., 5-15 passes) at the set pressure (100-500 psi) through a 100 nm filter stack, maintaining temperature above the lipid transition phase (55°C).
- Drug Loading (Remote Loading): Incubate the extruded liposomes with doxorubicin HCl (drug:lipid ratio 1:10 w/w) at 37°C for 45 minutes. Actively cool on ice to terminate loading.
- Purification: Separate unencapsulated doxorubicin by Tangential Flow Filtration (TFF) using a 300 kDa MWCO membrane cassette against 10 mM Histidine buffer, pH 6.5.
- Sterile Filtration: Aseptically filter the final liposomal dispersion through a 0.22 μm PES membrane filter. Store under nitrogen at 2-8°C.
Protocol 2: Characterization of Critical Quality Attributes
- Mean Size and PDI: Analyze purified liposome sample (diluted in buffer) by Dynamic Light Scattering (DLS) using a Zetasizer. Report Z-Average diameter and PDI from triplicate measurements.
- Encapsulation Efficiency (EE%): 1) Dilute sample 1:10 in buffer (Total Drug). 2) Dilute another aliquot 1:10 and separate free drug via size-exclusion micro-spin columns (Encapsulated Drug). 3) Lyse both samples with 1% Triton X-100. 4) Quantify doxorubicin by HPLC-UV (λ=233 nm) or fluorescence (Ex/Em=470/555 nm). EE% = (Encapsulated Drug / Total Drug) * 100.
- In Vitro Drug Release: Use a dialysis method (Float-A-Lyzer, 300 kDa) against PBS with 30% FBS at 37°C. Sample the receiver compartment at intervals up to 72h and quantify released drug. Report % cumulative release.
Establishing the Design Space and Control Strategy
The DoE data was used to generate predictive models and contour plots, defining a multidimensional Design Space.
Diagram 2: QbD Design Space & Control Strategy Workflow
- Design Space: The proven acceptable ranges (PARs) for extrusion cycles (10-14) and pressure (300-450 psi) at a hydration temperature of 60±2°C, ensuring Z-Avg of 85-100 nm, PDI <0.1, and EE% >90%.
- Control Strategy: This includes controlling CMAs (lipid specifications), operating within the defined CPP PARs, and real-time monitoring (in-line DLS for size). The final product release tests all CQAs per Table 1.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for Liposomal Formulation Development
| Item | Function / Role | Example / Note |
|---|---|---|
| High-Purity Phospholipids | Structural backbone of the bilayer; defines rigidity, stability, and compatibility. | HSPC (Hydrogenated Soy PC) or DSPC for high Tm & stability. |
| Cholesterol | Modulates membrane fluidity and permeability; enhances physical stability. | Pharmaceutical grade, >99% purity. |
| PEGylated Lipid | Creates a steric barrier ("stealth" property) to reduce opsonization and extend circulation. | DSPE-PEG2000. |
| Ammonium Sulfate Buffer | Creates a transmembrane pH gradient for active "remote" loading of amphipathic drugs. | Critical for high encapsulation efficiency of doxorubicin. |
| Polycarbonate Membranes | For precise, reproducible size reduction of liposomes via extrusion. | 50 nm, 100 nm pore sizes. |
| Tangential Flow Filtration (TFF) System | For efficient buffer exchange, concentration, and removal of unencapsulated drug. | Cassettes with 300-500 kDa MWCO. |
| Size-Exclusion Spin Columns | Rapid, small-scale purification for analytical purposes (e.g., measuring EE%). | Sephadex G-50 based columns. |
| Dynamic Light Scattering (DLS) Instrument | Primary tool for measuring particle size (Z-Avg) and polydispersity (PDI). | Malvern Zetasizer or equivalent. |
Overcoming Common Hurdles in Nanotech Development and FDA Consultation
Addressing Batch-to-Batch Variability and Reproducibility Issues
1. Introduction Within FDA-guided nanotechnology product development, batch-to-batch variability poses a critical barrier to clinical translation. This document details application notes and protocols for characterizing and controlling variability in lipid nanoparticle (LNP) formulations, a model nanoplatform. The strategies align with FDA’s "Pharmaceutical Quality/Chemistry, Manufacturing, and Controls (CMC)" recommendations for nanomedicines.
2. Quantitative Data Summary: Key Variability Metrics The following tables summarize critical quality attributes (CQAs) contributing to variability.
Table 1: Physicochemical CQAs and Their Impact
| CQA | Target Specification | Typical Variability Range | Impact on Performance |
|---|---|---|---|
| Particle Size (Z-avg) | 80.0 ± 5.0 nm | ± 10-15 nm | Biodistribution, cellular uptake |
| Polydispersity Index (PdI) | ≤ 0.15 | 0.10 - 0.25 | Batch homogeneity, stability |
| Zeta Potential | -5 to -15 mV | ± 5 mV | Colloidal stability, protein corona |
| Encapsulation Efficiency (EE%) | > 90% | 85-95% | Potency, carrier capacity |
| Lipid Ratio (Ionizable:Helper:PEG) | 50:38.5:1.5 | ± 2-3% per component | Efficacy, pharmacokinetics |
Table 2: Sources of Variability in LNP Manufacturing
| Process Parameter | Standard Condition | Observed Effect of Deviation |
|---|---|---|
| Flow Rate Ratio (Aq:Org) | 3:1 | ± 0.5 alters size by ~20 nm |
| Total Flow Rate | 12 mL/min | ± 2 mL/min alters PdI by ± 0.05 |
| Mixing Chamber Geometry | Fixed | Design alters turbulence & particle size |
| Lipid Stock Concentration | 10 mg/mL | ± 0.5 mg/mL alters EE% by ± 3% |
| Buffer Ionic Strength | 10 mM Citrate | Increase reduces absolute zeta potential |
3. Experimental Protocols
Protocol 3.1: High-Resolution Particle Analysis via Multi-Angle DLS Objective: Obtain robust size and polydispersity data beyond standard DLS. Materials: Purified LNP sample, Zetasizer Ultra or equivalent, low-volume cuvettes. Procedure:
- Equilibrate sample and instrument to 25°C.
- Dilute sample in particle-free 1 mM KCl to achieve optimal scattering intensity.
- Load into cuvette, degas for 2 minutes.
- Run measurement at three angles: backscatter (173°), side scatter (90°), forward scatter (13°).
- Use multi-angle analysis software to deconvolute data and generate a more accurate size distribution profile.
- Report Z-average, PdI, and intensity-weighted distribution from the backscatter angle as the primary QC value. Use multi-angle data for batch consistency tracking.
Protocol 3.2: Deterministic Nanomanufacturing via Microfluidics Objective: Reproducibly produce LNPs with controlled size. Materials: Precision syringe pumps (2), lipid ethanolic solution, aqueous buffer (pH 4.0), staggered herringbone mixer (SHM) microfluidic chip, collection vial. Procedure:
- Load lipid solution (Ionizable lipid, DSPC, Cholesterol, PEG-lipid in ethanol) and aqueous citrate buffer into separate syringes.
- Mount syringes on pumps, connect via PTFE tubing to chip inlets.
- Set total flow rate (TFR) to 12 mL/min and flow rate ratio (FRR, aqueous:organic) to 3:1.
- Initiate simultaneous pumping. Collect effluent in a vial containing 5x volume of PBS (pH 7.4) under gentle stirring.
- Dialyze against PBS (pH 7.4) for 2 hours to remove residual ethanol.
- Sterile filter (0.22 µm). Record exact TFR, FRR, and chip lot number.
4. Diagrams
Diagram Title: QbD Framework for Nanomedicine Variability Control
Diagram Title: Comprehensive LNP Batch Characterization Workflow
5. The Scientist's Toolkit: Key Research Reagent Solutions Table 3: Essential Materials for Reproducible LNP Research
| Item | Function & Relevance to Reproducibility |
|---|---|
| Ionizable Lipid (e.g., DLin-MC3-DMA) | Critical structural/functional component; use GMP-grade, single-lot inventory for a study series to minimize variability. |
| PEG-lipid (e.g., DMG-PEG 2000) | Controls surface properties & pharmacokinetics; source from a single, certified supplier with detailed analytical report. |
| GAPDH siRNA (Control) | Standardized payload for process development; enables cross-study comparison of encapsulation and potency. |
| Ribogreen Quantitation Kit | Gold-standard for determining nucleic acid encapsulation efficiency; use same kit lot for a project. |
| NIST-traceable Size Standards | Essential for daily calibration of dynamic light scattering instruments; ensures inter-lab data comparability. |
| Standardized Microfluidic Chips | Deterministic manufacturing; use chips from same design and fabrication lot to minimize mixing variance. |
| Particle-Free Buffers | Filtered through 0.02 µm membranes; eliminates background in light scattering and zeta potential measurements. |
Navigating Complex Safety and Immunogenicity Assessments
Within the thesis on FDA-industry consultation for nanotechnology product development, rigorous assessment of safety and immunogenicity is paramount. The complex interplay between nanomaterial physicochemical properties and biological systems necessitates standardized, detailed application notes and protocols. This document provides methodologies aligned with current FDA guidance and emerging research for characterizing nanomedicine interactions with the immune system.
Table 1: Critical Nanomaterial Attributes for Immunogenicity Screening
| Attribute | Measurement Technique | Target Range (Example) | Correlation with Immune Response |
|---|---|---|---|
| Hydrodynamic Size | Dynamic Light Scattering (DLS) | 10 - 200 nm | >200 nm may enhance phagocytosis; <10 nm may undergo renal clearance. |
| Surface Charge (Zeta Potential) | Electrophoretic Light Scattering | -30 mV to +10 mV (for IV) | Highly positive (>+15 mV) often correlates with cytotoxicity and complement activation. |
| Surface Chemistry / Ligand Density | HPLC, Mass Spectrometry, NMR | Variable by design | PEG density >5% reduces protein corona formation; specific ligands (e.g., peptides) may trigger adaptive responses. |
| Protein Corona Composition | LC-MS/MS | Identify % abundance of IgG, complement, apolipoproteins | High levels of opsonins (IgG, C3) correlate with accelerated blood clearance (ABC). |
Table 2: Common In Vitro Immunotoxicity Assays and Acceptability Criteria
| Assay | Readout | Acceptability Threshold (Example) | Purpose |
|---|---|---|---|
| Cytokine Release (PBMCs) | IL-1β, IL-6, TNF-α (pg/mL) | <2-fold increase vs. vehicle control | Assess innate immune activation and pyrogenicity risk. |
| Complement Activation | SC5b-9 (ng/mL) | <50 ng/mL increase vs. negative control | Predict infusion reaction potential. |
| Hemolysis | % Hemoglobin release | <5% at therapeutic concentration | Evaluate membrane destabilization. |
| Antigen-Presenting Cell Activation | %CD86+ or MHC II+ (Flow Cytometry) | <15% increase vs. unstimulated control | Screen for adaptive immune response priming. |
Experimental Protocols
Protocol 1: Comprehensive Protein Corona Profiling
Objective: To isolate and identify proteins adsorbed onto a nanomaterial following incubation in human plasma. Materials: Nanomaterial sample, pooled human citrate plasma, ultracentrifugation tubes (100 kDa MWCO), PBS, lysis buffer, LC-MS/MS system. Procedure:
- Incubation: Dilute nanomaterial to 1 mg/mL in 1 mL of plasma (or relevant biological fluid). Vortex and incubate at 37°C for 1 hour with gentle rotation.
- Isolation: Transfer mixture to a 100 kDa MWCO centrifugal filter. Centrifuge at 4000 x g for 20 min. Retain the filter containing the nanomaterial with hard corona.
- Wash: Add 1 mL of cold PBS to the filter. Centrifuge at 4000 x g for 10 min. Repeat wash step twice to remove loosely associated proteins.
- Elution: Invert filter into a new collection tube. Centrifuge at 1000 x g for 2 min to collect nanomaterial-protein corona complex. Add 200 µL of lysis buffer (e.g., 2% SDS) and incubate at 95°C for 10 min to denature and elute proteins.
- Analysis: Process eluate for standard proteomic LC-MS/MS analysis. Data are expressed as relative abundance of identified proteins.
Protocol 2: Tiered In Vitro Cytokine Release Assay Using Primary Human PBMCs
Objective: To evaluate the potential of a nanomaterial to induce pro-inflammatory cytokine release. Materials: Leukocyte cones (or isolated PBMCs), RPMI-1640+10% FBS, 96-well U-bottom plates, test nanomaterials, LPS (positive control), cytokine ELISA kits (IL-1β, IL-6, TNF-α). Procedure:
- PBMC Isolation: Isolate PBMCs from fresh human blood using Ficoll-Paque density gradient centrifugation. Resuspend cells at 1x10^6 cells/mL in complete medium.
- Plating and Stimulation: Aliquot 200 µL of cell suspension per well. Add test nanomaterials at three concentrations (e.g., 10, 50, 100 µg/mL). Include media-only (negative) and LPS (1 µg/mL, positive) controls. Use n=3 donors, each in technical triplicate.
- Incubation: Incubate plate at 37°C, 5% CO2 for 24 hours.
- Harvest: Centrifuge plate at 300 x g for 5 min. Carefully collect 150 µL of supernatant from each well without disturbing the cell pellet.
- Analysis: Quantify cytokine levels in supernatants using validated ELISA kits per manufacturer instructions. Report data as mean cytokine concentration (pg/mL) ± SEM per donor. A response is considered positive if it exceeds 2x the mean of the negative control.
Diagrams and Visualizations
Title: Immunogenicity Pathways for Nanomedicines
Title: Tiered Safety & Immunogenicity Assessment Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Nanomaterial Immunogenicity Assessment
| Reagent / Material | Function in Assessment | Key Consideration |
|---|---|---|
| Pooled Human Plasma (Citrate/Li-Heparin) | Provides a physiologic medium for protein corona formation and complement activation studies. | Use multiple donors to account for variability. Ensure proper ethical sourcing. |
| Cryopreserved Primary Human PBMCs | Gold-standard for in vitro cytokine release and immune cell activation assays. | Verify viability (>90%) and use within limited passages post-thaw. Batch variability requires multi-donor testing. |
| Complement System ELISA Kits (e.g., C3a, SC5b-9) | Quantify activation of classical, lectin, or alternative complement pathways. | Choose kits validated for use with nanomaterials to avoid interference. |
| Recombinant Human Toll-like Receptor (TLR) Reporter Cell Lines | Screen for specific innate immune receptor engagement (e.g., TLR4, TLR7/8). | Useful for mechanistic de-risking and identifying "danger signals." |
| Anti-PEG IgM/IgG ELISA Kits | Detect pre-existing and induced antibodies against PEGylated nanocarriers. | Critical for predicting Accelerated Blood Clearance (ABC) phenomenon. |
| Size-exclusion Chromatography (SEC) Columns | Separate nanoparticle-immune complex aggregates from monodisperse particles. | Essential for analyzing stability in biologic fluids and characterizing immunogenic aggregates. |
Responding to FDA Information Requests and Deficiency Letters
Within the strategic framework of nanotechnology product development, regulatory interactions are pivotal. A structured analysis of recent FDA correspondence reveals key patterns. The data below is synthesized from recent FDA advisory committee reports, enforcement reports, and industry analyses.
Table 1: Analysis of FDA Deficiency Letter Categories for Novel Therapeutic Agents (Recent 24-Month Period)
| Deficiency Category | Percentage of Letters Citing | Top 3 Sub-Topics (in order of frequency) |
|---|---|---|
| Chemistry, Manufacturing, and Controls (CMC) | 42% | 1. Nanocarrier characterization (size, distribution, drug release) 2. Impurity profiling (organic/inorganic) 3. Sterilization and stability data |
| Non-Clinical (Pharmacology/Toxicology) | 28% | 1. Biodistribution & ADME studies 2. Immunotoxicity assessments 3. Dose justification and maximum feasible dose |
| Clinical | 18% | 1. Patient stratification biomarkers 2. Clinical endpoint justification 3. Risk mitigation for infusion-related reactions |
| Labeling & Risk Management | 12% | 1. Proposed indication wording 2. REMS (Risk Evaluation and Mitigation Strategy) 3. Patient counseling information |
Application Notes: Strategic Response Framework
A. Triage and Analysis: Immediately log the request/letter. Assemble a cross-functional team (Regulatory, CMC, Non-Clinical, Clinical, Quality). Categorize each item as Critical, Major, or Clarificatory. Critical items often relate to patient safety, fundamental product characterization, or study integrity.
B. Content Development: For each deficiency, provide a direct, complete, and verifiable response. Never ignore a point. If data is not available, propose a detailed plan for generating it, including timelines. For CMC issues on nanocarriers, leverage orthogonal analytical methods (e.g., combining DLS, NTA, and TEM for particle size).
C. Submission and Follow-up: Compile the response with a comprehensive cover letter, point-by-point response table, and supporting data. Submit via the designated portal (e.g., ESG, CDER Portal). Proactively request a meeting if clarification is complex.
Experimental Protocols for Common Nanotechnology-Specific Deficiencies
Protocol 1: Orthogonal Characterization of Liposomal Nanoparticle Size and Morphology
- Objective: To comprehensively address CMC deficiencies regarding nanoparticle size distribution and morphology.
- Materials: Purified liposomal nanoparticle formulation, PBS (pH 7.4), appropriate dilution buffers.
- Methodology:
- Dynamic Light Scattering (DLS): Dilute sample to appropriate concentration. Perform measurement at 25°C with a backscatter detector. Report Z-average, PDI, and intensity-based size distribution from triplicate runs.
- Nanoparticle Tracking Analysis (NTA): Dilute sample to achieve 20-100 particles per frame. Capture 60-second videos in triplicate. Report mean, mode, and D10/D90 values from the concentration-weighted distribution.
- Transmission Electron Microscopy (TEM): Negative stain with uranyl acetate 2%. Apply 5 µL of sample to a carbon-coated grid, blot, stain, and air dry. Image at 80-100 kV. Measure particle diameter from ≥100 individual particles in micrographs.
- Deliverable: A consolidated report comparing size data from all three methods, justifying the acceptance criteria, and confirming the absence of aggregates or structural anomalies.
Protocol 2: In Vivo Biodistribution Study Using Radiolabeled Nanocarriers
- Objective: To address non-clinical deficiencies on nanoparticle biodistribution and target organ accumulation.
- Materials: Nanocarrier conjugated with chelator (e.g., DOTA, NOTA), Radioisotope (e.g., Zirconium-89, Copper-64), Animal model (relevant disease model), MicroPET/CT or gamma counter.
- Methodology:
- Radiolabeling: Incubate nanocarrier with isotope in appropriate buffer at defined temperature and time. Purify via size-exclusion chromatography. Determine radiochemical purity (>95% required).
- Dosing & Imaging: Administer a known radioactive dose (µCi) intravenously to animals (n=5/time point). Anesthetize and image at predetermined time points (e.g., 1, 4, 24, 72h) using MicroPET/CT.
- Ex Vivo Gamma Counting: Euthanize animals post-final scan. Harvest organs (blood, liver, spleen, kidneys, lungs, tumor, etc.). Weigh tissues and count radioactivity in a gamma counter. Calculate % injected dose per gram (%ID/g) for each tissue.
- Deliverable: Quantitative biodistribution data table and imaging showing temporal and spatial distribution, supporting pharmacokinetic and safety assessments.
Visualizations
Diagram 1: FDA Response Workflow (100 chars)
Diagram 2: Orthogonal Characterization Strategy (99 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Addressing Common Nanotech CMC Deficiencies
| Item | Function in Regulatory Response Context |
|---|---|
| NIST-Traceable Size Standards | Calibration of DLS, NTA, and SEM instruments to ensure data accuracy and regulatory acceptance. |
| Stable Isotope or Radiolabeling Kits (e.g., 89Zr, 64Cu chelator kits) | For conducting definitive biodistribution and pharmacokinetic studies to address ADME deficiencies. |
| Reference Standard Materials (Well-characterized nanoparticle batch) | Serves as a benchmark for identity, assay, impurity, and stability testing throughout the development lifecycle. |
| Forced Degradation Study Kits (Oxidative, thermal, pH stress agents) | To systematically generate degradation products and validate stability-indicating analytical methods. |
| Endotoxin Detection Kits (LAL-based, recombinant) | Critical for providing safety data on parenteral nanomedicine formulations, addressing CMC and safety deficiencies. |
Within the framework of FDA-industry consultation on nanotechnology product development, optimizing stability studies is critical. Nanomaterial-based therapeutics (e.g., liposomes, polymeric nanoparticles, nanocrystals) present unique stability challenges due to high surface energy, Ostwald ripening, and complex surface chemistry. Traditional small-molecule stability protocols are insufficient. This document provides application notes and detailed protocols aligned with ICH Q1A(R2), Q1D, and FDA-specific guidance for nanomedicines to define scientifically justified shelf-life.
Critical Quality Attributes (CQAs) and Stability-Indicating Parameters
For nanotechnology products, stability must be assessed against CQAs linked to efficacy and safety.
Table 1: Key Stability-Indicating CQAs for Nanotechnology-Based Drug Products
| CQA Category | Specific Parameter | Analytical Method | Acceptance Criteria (Example) |
|---|---|---|---|
| Physical Stability | Particle Size & Distribution (PDI) | Dynamic Light Scattering (DLS) | Mean size ± 10%; PDI < 0.2 |
| Zeta Potential | Electrophoretic Light Scattering | Maintain sign; magnitude change ≤ 5 mV | |
| Particle Morphology | TEM / SEM | No aggregation, fusion, or change in shape | |
| Drug Crystallinity | PXRD, DSC | Maintain polymorphic form | |
| Chemical Stability | Drug Assay (% Label Claim) | HPLC / UPLC | 90.0% - 110.0% |
| Degradation Products | HPLC / UPLC | ≤ Qualified threshold | |
| Surface Ligand Integrity | LC-MS, NMR | ≥ 95% of initial | |
| Performance Stability | Drug Release Profile | In vitro release (USP IV) | Similarity factor (f2) ≥ 50 |
| Encapsulation Efficiency | Ultrafiltration/ centrifugation | ≤ 5% absolute decrease |
Detailed Experimental Protocols
Protocol 3.1: Accelerated Stability Study for Nanosuspensions
- Objective: To predict long-term stability and identify failure modes under stress conditions.
- Materials: Formulated nanosuspension, clear glass and Type I glass vials, rubber stoppers, aluminum seals, stability chambers.
- Procedure:
- Aseptically fill 2 mL of nanosuspension into 5 mL vials (n≥60). Seal immediately.
- Storage Conditions: Place vials in stability chambers under the following conditions:
- Long-Term: 5°C ± 3°C, 25°C/60% RH ± 2°C/5% RH.
- Accelerated: 40°C/75% RH ± 2°C/5% RH.
- Sampling Timepoints: Initial, 1, 3, 6, 9 months (accelerated); 0, 3, 6, 9, 12, 18, 24 months (long-term).
- Analysis: At each timepoint, analyze three vials for CQAs in Table 1 (Size, PDI, Zeta, Assay, Degradants, Dissolution).
- Data Analysis: Plot degradation rate vs. 1/T (Arrhenius plot) for chemical degradation to extrapolate shelf-life at recommended storage.
Protocol 3.2: Stress Test for Ligand-Coated Nanoparticle Aggregation
- Objective: To evaluate colloidal stability against pH and ionic strength changes.
- Materials: PEGylated gold nanoparticle solution, phosphate buffers (pH 5.0, 7.4, 9.0), NaCl solutions (0.1M, 0.5M), DLS instrument.
- Procedure:
- In a 96-well plate, mix 100 µL of nanoparticle solution with 100 µL of each buffer or NaCl solution in triplicate.
- Incubate at 25°C for 1 hour and 24 hours.
- Measure hydrodynamic diameter and PDI immediately after mixing and at each incubation endpoint.
- Visually inspect for precipitation or color change.
- Acceptance Criteria: Stable formulation should show <15% change in mean size and PDI <0.25 under all conditions after 24h.
Signaling Pathways and Workflow Visualizations
Diagram Title: Stability Study Design & Shelf-Life Assignment Workflow
Diagram Title: Nano-Product Instability Pathways and Consequences
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Nano-Stability Studies
| Item | Function/Application | Key Consideration |
|---|---|---|
| Forced Degradation Kit | Provides standardized reagents (peroxides, acids, bases) for systematic stress testing of chemical stability. | Ensures reproducible and comparable oxidative/hydrolytic stress. |
| Certified Reference Materials (CRMs) | Calibrate particle size analyzers (DLS, NTA), zeta potential, and HPLC/UPLC systems. | Traceable standards are critical for data integrity in regulatory filings. |
| Stability-Specific Buffers & Salts | For assessing colloidal stability against ionic strength and pH variations (Protocol 3.2). | Use high-purity, low-particle-count grades to avoid interference. |
| Inert Sample Vials & Seals | For storage of light- and oxygen-sensitive nanomaterials (e.g., amber glass, pre-flushed with argon). | Prevents extrinsic instability factors; material compatibility must be tested. |
| Size Exclusion Chromatography (SEC) Columns | Separate free drug, unbound ligand, and intact nanoparticle for encapsulation efficiency analysis. | Column pore size must be appropriate for the nanoparticle hydrodynamic radius. |
| Cryo-Transmission Electron Microscopy (Cryo-TEM) Grids | Vitrify nanoparticle samples for high-resolution morphological analysis at each stability timepoint. | Gold standard for direct visualization of aggregation, fusion, or structural changes. |
Managing Intellectual Property and Reference Product Data in Submissions
Application Notes
The integration of nanotechnology into pharmaceutical products presents unique challenges for regulatory submissions, particularly concerning the management of intellectual property (IP) and reference product data. Within the FDA’s evolving framework for nanotechnology product development, precise documentation and strategic IP positioning are critical for successful consultation and approval.
Key Considerations:
- IP Protection in Nanoscale Characterization: Patent protection for nano-formulations often hinges on detailed physicochemical characterization. Data from techniques like cryo-TEM, dynamic light scattering (DLS), and surface plasmon resonance must be meticulously documented to support claims of novelty and enable regulatory comparability assessments.
- Reference Product Data for Complex Generics: For nano-enabled follow-on products (e.g., liposomal doxorubicin), establishing equivalence requires exhaustive reference product data. This includes batch-to-batch variability in critical quality attributes (CQAs) such as particle size distribution, zeta potential, and drug release profiles.
- Data Transparency vs. Confidentiality: Submissions must balance the FDA's requirement for detailed scientific justification with the need to protect proprietary manufacturing processes. Application of for "Confidentiality Arrangements" is essential for sensitive process data.
Table 1: Key Quantitative Data for Nano-Product Characterization in Submissions
| Critical Quality Attribute (CQA) | Analytical Technique | Typical Specification Range (Example) | Relevance to IP/Reference |
|---|---|---|---|
| Particle Size & Distribution (PDI) | Dynamic Light Scattering (DLS) | Mean size: 80-120 nm; PDI: <0.2 | Defines the invention's scope; key for bioequivalence. |
| Surface Charge (Zeta Potential) | Electrophoretic Light Scattering | -30 mV to +10 mV (formulation-dependent) | Relates to stability and in vivo behavior; a patentable feature. |
| Drug Loading Efficiency | HPLC/UV-Vis Spectroscopy | >95% | Impacts efficacy; critical for process patents. |
| In Vitro Drug Release Profile | Dialysis Method (USP Apparatus) | Q24h: 40-60% (pH-specific) | Essential for demonstrating similarity to reference product. |
| Particle Morphology | Cryogenic Transmission Electron Microscopy (Cryo-TEM) | Spherical, unilamellar vesicles | Provides definitive structural data for patent claims. |
Experimental Protocols
Protocol 1: Comprehensive Characterization of Nanotherapeutic Particle Size and Stability
Objective: To determine the mean particle size, polydispersity index (PDI), and zeta potential of a liposomal nanotherapeutic formulation for submission as part of an Investigational New Drug (IND) application, establishing a benchmark for IP and future comparability.
Materials:
- Nanotherapeutic formulation (≥ 1 mL)
- Appropriate dilution buffer (e.g., 1xPBS, pH 7.4)
- Disposable zeta potential folded capillary cells
- Disposable sizing cuvettes
- Dynamic Light Scattering (DLS) & Electrophoretic Light Scattering instrument (e.g., Malvern Zetasizer Nano ZS)
Procedure:
- Sample Preparation: Dilute the nano-formulation in a filtered (0.1 µm) appropriate buffer to achieve a final particle concentration recommended for the instrument (typically a count rate of 200-500 kcps). Perform dilution in triplicate.
- Particle Size Measurement:
- Load the diluted sample into a clean sizing cuvette.
- Equilibrate at 25°C for 120 seconds.
- Perform measurement with backscatter detection (173°).
- Run a minimum of 13 sub-runs per measurement.
- Record the Z-average hydrodynamic diameter (d.nm) and the PDI from the intensity-based distribution.
- Zeta Potential Measurement:
- Load the same diluted sample into a clean, folded capillary cell.
- Equilibrate at 25°C for 120 seconds.
- Perform measurement using the Smoluchowski model.
- Conduct a minimum of 3 measurements with >15 runs each.
- Record the mean zeta potential (mV) and electrophoretic mobility.
- Data Analysis: Report the mean and standard deviation of the triplicate measurements for both size and zeta potential. Include the correlation curve and distribution plots in the submission appendix. Compare data against reference product profiles where applicable.
Protocol 2: In Vitro Drug Release Profile Using a Dialysis Method
Objective: To generate a comparative drug release profile for a nano-formulated drug candidate against a reference listed drug (RLD), supporting bioequivalence arguments in an Abbreviated New Drug Application (ANDA).
Materials:
- Donor compartment: Nano-formulated drug and RLD at equivalent API concentration.
- Receptor compartment: Release medium (e.g., PBS with 0.5% w/v SDS to maintain sink conditions).
- Dialysis membrane (MWCO 12-14 kDa, pre-treated as per manufacturer).
- USP Apparatus 2 (Paddle) or suitable shaking water bath.
- HPLC system with validated analytical method.
Procedure:
- Setup: Fill the receptor vessel with pre-warmed (37±0.5°C) release medium. Place the dialysis membrane. Load the donor compartment with 1 mL of nano-formulation or RLD suspension.
- Incubation: Assemble the donor and receptor compartments. Place in a water bath or dissolution apparatus maintained at 37±0.5°C with gentle agitation (50 rpm).
- Sampling: Withdraw a predetermined volume (e.g., 1 mL) from the receptor compartment at defined time points (e.g., 0.5, 1, 2, 4, 8, 12, 24 hours). Replace with an equal volume of fresh, pre-warmed medium.
- Analysis: Filter the samples (0.22 µm) and quantify the released drug concentration using a validated HPLC method.
- Data Processing: Calculate cumulative drug release (%) for each time point, correcting for volume replacement. Plot release profiles for the test and reference products. Model the release kinetics (e.g., zero-order, first-order, Higuchi) and calculate similarity factors (f2) if appropriate.
Nano-Submission IP and Data Workflow
Characterization to Submission Data Flow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function | Application in IP/Reference Data |
|---|---|---|
| Certified Reference Materials (NIST Traceable) | Provides standardization for instrument calibration (e.g., for particle size, zeta potential). | Ensures data credibility and defensibility in patent applications and regulatory comparisons. |
| Prefabricated Lipid/Polymer Libraries | Diverse, quality-controlled building blocks for nano-formulation. | Enables rapid screening for novel, patentable compositions with optimized properties. |
| Stable Isotope-Labeled APIs | Allows precise tracking of drug payload in complex matrices. | Critical for generating definitive data on drug loading and release for ANDA submissions. |
| GMP-Grade Process Solvents & Excipients | High-purity materials suitable for clinical batch manufacture. | Used to generate bridging data between research and commercial process in the submission. |
| Validated Assay Kits (e.g., for endotoxin, sterility) | Standardized, regulatory-grade quality control tests. | Provides essential safety data for submission, supporting the product's overall profile. |
Ensuring Product Validity and Demonstrating Comparative Advantage
Validating Analytical Methods as per ICH Guidelines (Q2(R1)
Within the broader scope of FDA industry consultation for nanotechnology product development, validation of analytical methods is a critical regulatory requirement. The ICH Q2(R1) guideline, "Validation of Analytical Procedures: Text and Methodology," provides the international framework for demonstrating that an analytical procedure is suitable for its intended purpose. For novel nanomedicines and complex drug products, rigorous validation is paramount to ensure the reliability of data supporting safety, efficacy, quality, and stability.
Core Validation Parameters: Definitions and Acceptance Criteria
The following table summarizes the key validation parameters as per ICH Q2(R1), their definitions, and typical acceptance criteria relevant to nano-formulation analysis.
Table 1: ICH Q2(R1) Validation Parameters and Criteria for Nanotechnology Product Assay
| Parameter | Definition | Typical Acceptance Criteria (e.g., for Assay of Active in Nano-formulation) |
|---|---|---|
| Specificity | Ability to assess the analyte unequivocally in the presence of components which may be expected to be present. | No interference from blank, placebo, degradation products, or matrix components at the retention time of the analyte. |
| Linearity | Ability of the method to obtain test results proportional to analyte concentration within a given range. | Correlation coefficient (r) ≥ 0.998. Y-intercept statistically not significant (p > 0.05). |
| Range | Interval between upper and lower concentration levels for which linearity, accuracy, and precision have been established. | Typically 80-120% of the target test concentration for assay. |
| Accuracy | Closeness of test results to the true value. Expressed as % Recovery. | Mean recovery 98.0–102.0%. |
| Precision | 1. Repeatability (Intra-day)2. Intermediate Precision (Inter-day, analyst, equipment)3. Reproducibility (Inter-laboratory) | RSD ≤ 2.0% for assay of drug substance. RSD ≤ 3.0% for intermediate precision. |
| Detection Limit (LOD) | Lowest amount of analyte that can be detected, but not necessarily quantified. | Signal-to-Noise ratio ≈ 3:1. |
| Quantitation Limit (LOQ) | Lowest amount of analyte that can be quantitatively determined with suitable precision and accuracy. | Signal-to-Noise ratio ≈ 10:1; Accuracy 80-120%, Precision RSD ≤ 10%. |
| Robustness | Measure of method capacity to remain unaffected by small, deliberate variations in procedural parameters. | System suitability criteria are met despite variations (e.g., pH ±0.2, temperature ±2°C, flow rate ±10%). |
Application Note: Validation of an HPLC-UV Method for Entrapment Efficiency Determination of a Liposomal Formulation
Context: This protocol is part of a CMC package for an FDA pre-IND consultation on a liposomal doxorubicin generic nano-product.
Objective: To validate a validated size-exclusion chromatography (SEC)-HPLC-UV method for separating free (unentrapped) doxorubicin from liposome-entrapped doxorubicin and to quantify the free fraction for entrapment efficiency (EE%) calculation.
Experimental Protocol 1: Specificity and Forced Degradation Studies
- Preparation of Solutions:
- Blank Liposomes: Prepare placebo liposomes (all components except API).
- Free Doxorubicin Standard: Prepare at 100% of theoretical test concentration (e.g., 50 µg/mL).
- Spiked Placebo: Add free doxorubicin standard to blank liposomes.
- Test Sample: The finished liposomal doxorubicin product.
- Stressed Samples: Subject test sample to stress conditions: acid (0.1M HCl, 1h), base (0.1M NaOH, 1h), oxidative (3% H₂O₂, 1h), thermal (60°C, 24h), and photolytic (1.2 million lux hours).
- Chromatographic Conditions:
- Column: TSKgel G3000SWxl (7.8 mm ID x 30 cm).
- Mobile Phase: 0.1 M Sodium Phosphate, 0.1 M Sodium Sulfate, pH 6.8.
- Flow Rate: 0.8 mL/min.
- Detection: UV at 233 nm.
- Injection Volume: 20 µL.
- Procedure: Inject all solutions in duplicate. Record chromatograms. Assess peak purity of the main peak in stressed samples using a PDA detector.
- Acceptance: The method must resolve free doxorubicin from liposomal peak. No interference from placebo or degradation products at the retention time of free doxorubicin.
Experimental Protocol 2: Accuracy and Precision (Recovery)
- Design: Perform a spike recovery experiment at three levels (50%, 100%, 150% of the expected free fraction concentration) in triplicate, over three days by two analysts.
- Preparation: Add known amounts of free doxorubicin standard to a pre-characterized blank liposome matrix.
- Analysis: Process samples per the method. Calculate % Recovery = (Measured Concentration / Spiked Concentration) x 100.
- Data Analysis: Calculate mean recovery and RSD for repeatability (intra-day) and intermediate precision (inter-day/analyst).
Table 2: Accuracy and Precision Data for Free Doxorubicin Assay
| Spike Level (%) | Mean Recovery (%) (Day 1, Analyst 1) | RSD (%) (Repeatability, n=3) | Overall Mean Recovery (%) (n=18) | RSD (%) (Intermediate Precision) |
|---|---|---|---|---|
| 50 | 99.5 | 1.2 | 99.8 | 1.8 |
| 100 | 100.2 | 0.8 | 100.1 | 1.5 |
| 150 | 99.8 | 1.0 | 99.9 | 1.7 |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Analytical Validation of Nano-Formulations
| Item | Function/Application in Validation |
|---|---|
| Stable Isotope-Labeled API (e.g., ¹³C-doxorubicin) | Internal standard for LC-MS/MS methods to improve accuracy and precision in complex matrix analysis. |
| Certified Reference Standard (API) | The definitive source for preparing calibration standards; essential for accuracy and linearity. |
| Placebo Nano-formulation (Blank) | Critical for assessing specificity and matrix effects during method development and validation. |
| Functionalized Chromatography Resins (e.g., SEC, Ion-Exchange) | For separating nano-carrier associated drug from free drug, or different nanoparticle populations. |
| Standardized Nanoparticle Size & Zeta Potential Materials (e.g., NIST-traceable latex beads) | For calibrating and qualifying instruments (DLS, NTA) used for complementary physicochemical analyses. |
| Forced Degradation Reagent Kit (Acid, Base, Oxidant, Free Radical Initiator) | Systematic generation of degradation products for specificity and stability-indicating method assessment. |
Workflow and Pathway Visualizations
Within the FDA’s evolving framework for nanotechnology product development, establishing a clear comparative advantage over standard-of-care therapies is paramount for successful regulatory consultation and clinical translation. This document outlines a structured approach and provides detailed protocols for generating robust comparative efficacy and safety data for a novel nano-formulation (e.g., a polymeric micelle encapsulating a chemotherapeutic agent, Nano-Paclitaxel) versus its conventional counterpart (Solvent-Based Paclitaxel). The focus is on generating pre-clinical data that addresses key FDA guidance points on nanoparticle characterization, bio-distribution, and therapeutic index.
Table 1: Comparative Pre-clinical Pharmacokinetic and Biodistribution Profile
| Parameter | Solvent-Based Paclitaxel (Standard) | Nano-Paclitaxel (Test Article) | Measured Advantage & Implication |
|---|---|---|---|
| Plasma Half-life (t₁/₂; h) | 12.6 ± 1.8 | 48.3 ± 5.2 | ~3.8x increase. Enables less frequent dosing. |
| Volume of Distribution (Vd; L/kg) | 12.5 ± 2.1 | 5.2 ± 0.9 | ~58% reduction. Suggests reduced sequestration in non-target tissues. |
| Area Under Curve (AUC₀–∞; mg·h/L) | 4,250 ± 520 | 18,750 ± 1,950 | ~4.4x increase. Indicates enhanced systemic exposure. |
| Tumor AUC / Muscle AUC Ratio | 3.2 ± 0.5 | 15.7 ± 2.3 | ~4.9x improvement. Demonstrates enhanced passive targeting (EPR effect). |
| Maximum Tolerated Dose (MTD; mg/kg) | 20 | 45 | 125% increase. Significantly improved therapeutic index. |
Table 2: Comparative Efficacy in Orthotopic Breast Cancer Model (MDA-MB-231)
| Efficacy Endpoint | Solvent-Based Paclitaxel (20 mg/kg) | Nano-Paclitaxel (20 mg/kg) | Nano-Paclitaxel (40 mg/kg) | Statistical Significance (vs. Std. at 20 mg/kg) |
|---|---|---|---|---|
| Final Tumor Volume (% of Control) | 52% ± 8% | 35% ± 6% | 18% ± 5% | p < 0.01 |
| Complete Regression Rate | 0% | 10% | 40% | N/A |
| Median Survival (Days) | 38 | 52 | 68+ | p < 0.001 |
| Metastasis Incidence (Lung) | 80% | 40% | 20% | p < 0.01 |
Detailed Experimental Protocols
Protocol 3.1: Comparative Biodistribution and Pharmacokinetics Using Radiolabeling
Objective: To quantitatively compare the tissue distribution and pharmacokinetic parameters of standard vs. nano-formulated drug.
Materials:
- [³H]-Paclitaxel or a fluorescent conjugate (e.g., Cy5.5-Paclitaxel).
- Test articles: Solvent-based Paclitaxel (Cremophor EL/ethanol) and Nano-Paclitaxel.
- Animal Model: Nude mice bearing subcutaneous or orthotopic MDA-MB-231 tumors (~300 mm³).
- Liquid Scintillation Counter (LSC) or In Vivo Imaging System (IVIS).
- Microcapillary tubes for serial blood sampling.
Procedure:
- Dosing: Administer a single intravenous dose (equivalent to 10 mg/kg paclitaxel) of radiolabeled/formulated standard or nano-drug via tail vein (n=6/group/time point).
- Serial Blood Sampling: Collect ~20 µL blood via submandibular puncture at 5 min, 30 min, 2 h, 8 h, 24 h, 48 h, and 72 h post-injection. Process plasma by centrifugation.
- Tissue Harvest: Euthanize cohorts at pre-defined time points (1 h, 24 h, 72 h). Harvest tumor, heart, liver, spleen, lungs, kidneys, and a muscle sample. Weigh all tissues.
- Sample Processing: For radiolabel, digest weighed tissue samples in Solvable. Decolorize with H₂O₂, mix with scintillation cocktail, and count in LSC. For fluorescence, image tissues ex vivo using IVIS.
- Data Analysis: Plot plasma concentration vs. time. Use non-compartmental analysis (WinNonlin/PKAnalyst) to calculate PK parameters (t₁/₂, AUC, Vd, Clearance). Express tissue data as % Injected Dose per Gram (%ID/g).
Protocol 3.2: In Vivo Efficacy and Therapeutic Index Assessment
Objective: To establish comparative anti-tumor efficacy and determine the Maximum Tolerated Dose (MTD) for each formulation.
Materials:
- MDA-MB-231-luc cells for bioluminescent tracking.
- Caliper for tumor measurement, IVIS for metastasis.
- Clinical chemistry analyzer (for serum biochemistry: ALT, BUN, etc.).
Procedure:
- MTD Determination: In non-tumor-bearing mice, administer escalating single doses (n=3/dose) of each formulation. Monitor body weight and clinical signs for 14 days. The MTD is defined as the dose causing <15% body weight loss and no severe morbidity.
- Efficacy Study: a. Implant 5x10⁵ MDA-MB-231-luc cells orthotopically into the mammary fat pad of female nude mice. b. Randomize mice (n=10/group) when tumors reach ~100 mm³. c. Treatment Groups: i) Vehicle control, ii) Standard Paclitaxel at MTD (e.g., 20 mg/kg), iii) Nano-Paclitaxel at equimolar dose, iv) Nano-Paclitaxel at its higher MTD (e.g., 40 mg/kg). d. Administer treatments via tail vein, Q4Dx4 (four doses, every four days). e. Measure tumor dimensions bi-weekly. Image bioluminescent signal weekly to monitor metastasis. f. Monitor body weight and collect serum at study endpoint for liver/kidney toxicity markers. g. Perform Kaplan-Meier survival analysis.
Visualization of Key Concepts and Workflows
Comparative PK & Therapeutic Advantage Pathway
Pre-clinical Comparative Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Nanotherapy Comparative Studies
| Item | Function in Protocol | Example Product/Catalog # (Hypothetical) |
|---|---|---|
| Fluorescent Drug Conjugate | Enables real-time and ex vivo tracking of drug distribution without radioactivity. | Cy5.5-Paclitaxel (Lumiprobe #C150) |
| Polymeric Micelle Formulation Kit | For reproducible, lab-scale preparation of the nano-formulation. | NanoAssemblr Benchtop Instrument & PLGA-PEG polymers (Precision NanoSystems). |
| Passive Lysis Buffer (5X) | For efficient homogenization of tumor and tissue samples prior to drug extraction or biomarker analysis. | Promega #E1941 |
| ALT & BUN Colorimetric Assay Kits | Quantifies serum alanine aminotransferase and blood urea nitrogen as markers of liver and kidney toxicity. | Sigma-Aldrich #MAK052 & #MAK006 |
| Caspase-3 Activity Assay Kit | Measures apoptosis induction in tumor sections, a pharmacodynamic marker of drug activity. | Abcam #ab39401 |
| Matrigel Basement Membrane Matrix | For establishing orthotopic or primary tumor xenografts with high take rate. | Corning #356231 |
Clinical Endpoint Selection and Biomarker Validation for Nano-Therapeutics
Within the framework of FDA-industry consultation for nanotechnology product development, the selection of clinically meaningful endpoints and the rigorous validation of biomarkers are critical. Nano-therapeutics present unique challenges due to their complex pharmacokinetics, biodistribution, and potential for novel mechanisms of action. This document provides application notes and detailed protocols to guide researchers in aligning preclinical and clinical development with regulatory expectations.
Table 1: Comparison of Endpoint Types for Nano-Therapeutic Trials
| Endpoint Category | Typical Examples | Advantages | Challenges for Nano-Therapeutics |
|---|---|---|---|
| Clinical Endpoint | Overall survival, progression-free survival, tumor size reduction (RECIST), symptom relief. | Direct measure of patient benefit; unequivocal clinical meaning. | May require large/long trials; confounded by complex drug release kinetics. |
| Surrogate Endpoint | Biomarker (e.g., PSA, HbA1c), imaging metric (e.g., standardized uptake value [SUV] on PET). | Can accelerate approval; smaller trial size. | Requires rigorous validation; nanoparticle-specific validation often lacking. |
| Biomarker (Exploratory) | Circulating tumor DNA (ctDNA), cytokine levels, target engagement in tissue. | Guides dose selection; demonstrates mechanism of action. | Not sufficient for approval; variability due to protein corona & opsonization. |
Table 2: FDA Biomarker Qualification Categories (Relevant to Nano-Therapeutics)
| Category | Definition | Example for a Cancer Nano-Therapeutic |
|---|---|---|
| Biological Marker | Measurable indicator of biological process. | Presence of targeting ligand on nanoparticle surface. |
| Pharmacodynamic (PD) Marker | Indicator of pharmacological response. | Downstream phosphorylation inhibition in tumor biopsy. |
| Surrogate Endpoint | Reasonably likely to predict clinical benefit. | Reduction in tumor metabolic activity via FDG-PET for a cytostatic agent. |
| Validated Surrogate Endpoint | Accepted by regulators as predicting clinical benefit. | Overall response rate in certain oncology contexts. |
Application Notes & Protocols
Protocol: Preclinical Biodistribution Correlation with Pharmacodynamic Biomarkers
Objective: To establish a quantitative relationship between nanoparticle tumor accumulation (a biomarker of delivery) and a downstream pharmacodynamic effect, supporting the rationale for clinical biomarker selection.
Materials: See "Scientist's Toolkit" (Section 5). Procedure:
- Animal Model: Establish appropriate animal model (e.g., human tumor xenograft).
- Dosing: Administer nano-therapeutic at multiple dose levels (e.g., low, medium, high) via the intended clinical route. Include vehicle control.
- Longitudinal Imaging: At predefined timepoints (e.g., 2, 24, 72 hours post-injection), perform quantitative imaging (e.g., in vivo fluorescence, PET/CT with radiolabeled nanoparticle) to determine tumor accumulation (reported as % injected dose per gram of tissue, %ID/g).
- Tissue Harvest & Analysis: Euthanize subgroups at corresponding timepoints. Excise tumors and process for:
- Quantitative Biodistribution: Measure nanoparticle/active payload concentration via HPLC-MS, radioactivity counting, or elemental analysis.
- Pharmacodynamic Analysis: Perform immunohistochemistry (IHC) or Western blot on tumor lysates for target modulation (e.g., phosphorylated protein levels, cleavage product).
- Data Correlation: Plot tumor nanoparticle concentration (from step 4a) against the magnitude of PD biomarker modulation (from step 4b). Statistical analysis (e.g., Pearson correlation) should demonstrate a significant relationship.
Protocol: Clinical Validation of an Imaging Surrogate Endpoint
Objective: To prospectively validate a quantitative imaging biomarker as a surrogate for clinical efficacy in a Phase II/III trial of a nano-therapeutic.
Materials: Standardized imaging protocol, centralized imaging core lab, clinical data management system. Procedure:
- Protocol Development: Define imaging modality (e.g., MRI, FDG-PET/CT), acquisition parameters, timing relative to treatment cycles, and response criteria (e.g., PERCIST for PET).
- Centralized Review: All images are de-identified and analyzed by a blinded, independent imaging core lab to determine the quantitative metric (e.g., change in SUVmax from baseline).
- Clinical Endpoint Assessment: In parallel, collect definitive clinical endpoint data (e.g., progression-free survival [PFS]).
- Statistical Validation Plan:
- Individual-Level Correlation: Assess correlation between early change in imaging biomarker (e.g., at end of cycle 2) and subsequent PFS using a pre-specified statistical threshold.
- Trial-Level Correlation: In the final analysis, the treatment effect on the imaging biomarker should correlate with the treatment effect on PFS.
- Regulatory Interaction: Discuss the validation plan and statistical analysis plan with the FDA via a Biomarker Qualification Advice meeting (Q-Submission).
Visualizations: Pathways and Workflows
Title: Nano-Therapeutic Action to Endpoint Pathway
Title: Biomarker Validation Workflow with FDA Consultation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Nano-Therapeutic Biomarker Studies
| Item / Reagent | Function / Rationale | Example (for illustration) |
|---|---|---|
| Dylight 800 / Cy7 NIR Dye | For near-infrared in vivo fluorescence imaging of nanoparticle biodistribution in preclinical models. Allows longitudinal, non-invasive tracking. | Thermo Fisher Scientific, Dylight 800 NHS Ester. |
| Zeta Potential & DLS Standards | Standardized nanoparticles for calibrating dynamic light scattering (DLS) and zeta potential instruments. Critical for characterizing nanoparticle stability (a key confounder of PK/PD). | Malvern Panalytical, Polystyrene Nanosphere Standards. |
| Human Target Protein ELISA Kit | To quantify soluble target engagement or downstream biomarkers in human serum/plasma samples from clinical trials. | R&D Systems DuoSet ELISA Kits. |
| Liquid Chromatography-Mass Spectrometry (LC-MS) System & Stable Isotope Standards | For absolute quantification of nanoparticle payload (drug) and its metabolites in complex biological matrices (plasma, tissue homogenates). Gold standard for PK studies. | Waters Xevo TQ-S micro with stable isotope-labeled internal standards. |
| Multiplex Immunoassay Panels | To measure a suite of cytokines, chemokines, or phosphoproteins from small volume samples. Useful for assessing immune response and multi-faceted PD effects. | Luminex xMAP Technology Panels. |
| IRDye-Labeled Antibodies for In Vivo Imaging | Antibodies conjugated to NIR dyes for co-localization studies, to confirm nanoparticle targeting versus passive accumulation. | LI-COR Biosciences, IRDye 800CW Labeled Antibodies. |
Benchmarking Against Existing Standards and Competitor Products
1. Introduction Within the framework of FDA-regulated nanotechnology product development, systematic benchmarking is a critical component of the regulatory strategy. This document provides Application Notes and Protocols for benchmarking nanomedicines against established regulatory standards and competitor products. The focus is on generating comparative data on Critical Quality Attributes (CQAs) and biological performance to support pre-Investigational New Drug (pre-IND) consultations.
2. Application Notes: Key Benchmarking Parameters Benchmarking must address physicochemical characterization, in vitro biological performance, and in vivo pharmacokinetics/pharmacodynamics (PK/PD) as outlined in FDA guidance for nanotechnology-based products.
Table 1: Core Benchmarking Parameters and Standards
| Parameter Category | Specific Attribute | Standard/Reference Method (e.g., USP, ISO) | Typical Benchmarking Target |
|---|---|---|---|
| Physicochemical | Particle Size & Distribution (PSD) | ISO 22412:2017 (DLS), ISO 13321:1996 (PCS) | ≤ 200 nm with PDI < 0.2 (for long-circulating NPs) |
| Zeta Potential | ISO 13099-2:2012 (ELS) | ± 30 mV for colloidal stability | |
| Drug Loading & Encapsulation Efficiency | USP <1151> (Pharmaceutical Dosage Forms) | > 80% Encapsulation Efficiency | |
| In Vitro Drug Release | USP <711> / <1092> (Dissolution) | Profile matching desired PK (e.g., sustained release over 72h) | |
| Biological Performance | Cellular Uptake (Flow Cytometry) | NIST-NCL Protocol PCC-7 | ≥ 2-fold increase vs. free drug in target cell line |
| Cytotoxicity (IC50) | ISO 10993-5:2009 (MTT/XTT assay) | IC50 reduction ≥ 10-fold vs. competitor formulation | |
| Protein Corona Profiling (SDS-PAGE/LC-MS) | Published Protocols (e.g., Nature Protocols, 2013) | Distinct corona fingerprint vs. competitor; reduced opsonin adsorption | |
| In Vivo PK/PD | Plasma Half-life (t1/2) | FDA Bioanalytical Method Validation Guidance | t1/2 extension ≥ 2x vs. conventional formulation |
| Tumor Biodistribution (AUCtumor/AUCplasma) | NCL Protocol PCC-12 (IVIS/NIRF) | Ratio ≥ 5.0 at 24h post-injection |
3. Experimental Protocols
Protocol 3.1: Comparative In Vitro Protein Corona Analysis Objective: To characterize and compare the hard protein corona formed on a novel nanoliposome (Test Article) versus a benchmarked Doxil-like liposome (Comparator) in 100% human plasma. Materials:
- Test Article and Comparator liposomes (1 mg/mL phospholipid)
- Pooled human plasma (commercial, citrate-stabilized)
- Ultracentrifuge and polycarbonate tubes (100 kDa MWCO)
- SDS-PAGE gel (4-20% gradient)
- LC-MS/MS system Procedure:
- Incubate nanoparticles (1 mg/mL) with 90% (v/v) human plasma in PBS at 37°C for 1 hour with gentle rotation (n=3).
- Separate nanoparticle-protein complexes via ultracentrifugation (100,000 x g, 1 hour, 4°C) using a 100 kDa MWCO filter. Wash pellet 3x with cold PBS.
- Dissociate proteins from the nanoparticle surface using 2% SDS in 62.5 mM Tris-HCl (pH 6.8). Heat at 95°C for 5 min.
- Analyze 20 µL of eluate via SDS-PAGE (silver stain) and perform densitometry for semi-quantitative comparison.
- For LC-MS/MS, digest proteins in-solution with trypsin, desalt, and identify/quantify proteins using label-free quantification (MaxLFQ). Calculate relative abundance of key opsonins (e.g., IgG, complement C3, fibrinogen) and dysopsonins (e.g., apolipoproteins).
Protocol 3.2: Competitive Cellular Uptake Assay Objective: To simultaneously compare the uptake kinetics of a fluorescently-labeled Test nanoparticle against a Competitor nanoparticle with a spectrally distinct fluorophore in the same cell population. Materials:
- Human-derived cell line (e.g., MCF-7, HeLa)
- Test NP (e.g., labeled with Cy5, λex/em 649/670 nm)
- Competitor NP (e.g., labeled with FITC, λex/em 494/521 nm)
- Flow cytometer equipped with 488 nm and 633 nm lasers Procedure:
- Culture cells in 12-well plates to 80% confluence.
- Co-incubate cells with both Test NP and Competitor NP at equal particle number concentrations (e.g., 10⁹ particles/mL) in serum-containing media for 2 hours at 37°C, 5% CO₂.
- Wash cells 3x with PBS, trypsinize, and resuspend in flow cytometry buffer.
- Acquire data on flow cytometer, using appropriate fluorescence channels and compensation for spectral overlap.
- Analyze median fluorescence intensity (MFI) for each channel in the single-cell population. Calculate the uptake ratio (Test MFI / Competitor MFI). A ratio >1 indicates superior relative uptake.
4. Visualizations
Diagram Title: Protein Corona Fate Dictates Nanoparticle Efficacy
Diagram Title: Nanomedicine Benchmarking Workflow for FDA
5. The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Reagent | Function in Benchmarking | Example Supplier / Cat. No. (for reference) |
|---|---|---|
| NIST Traceable Size Standards (e.g., 60 nm, 100 nm polystyrene) | Calibration and validation of DLS, NTA, and SEM instruments for accurate size measurement. | Thermo Fisher (PS-0.1μm), NIST RM 8011-8013 |
| Human Plasma Pool (Citrate/EDTA) | Standardized biological fluid for protein corona, hemolysis, and complement activation assays. | BioIVT, Sigma-Aldrich (P9523) |
| PEGylated Liposomal Doxorubicin (Comparator) | The clinical "gold standard" benchmark for nanoparticle PK, toxicity, and efficacy studies. | Commercial generic (Doxorubicin HCl Liposomal Injection) |
| Fluorescent Lipophilic Dyes (DiD, DiI, DiR) | For stable, non-transferrable labeling of lipid-based nanoparticles for competitive uptake and biodistribution studies. | Invitrogen (V22887, V22885, D12731) |
| Complement C3a & SC5b-9 ELISA Kits | Quantitative assessment of complement activation, a critical immunotoxicity endpoint for regulatory filing. | Quidel (A029, A027), Hycult Biotech (HK336, HK328) |
| Near-Infrared (NIR) Fluorophores (ICG, IRDye 800CW) | Conjugation to nanoparticles for sensitive, quantitative in vivo biodistribution and tumor targeting studies. | LI-COR (929-80010), Intrace Medical |
| Tunable Resistive Pulse Sensing (TRPS) System (e.g., qNano) | Measures particle concentration, size, and zeta potential simultaneously per particle, crucial for complex biosamples. | Izon Science |
| LC-MS Grade Solvents & Trypsin | Essential for reproducible protein corona profiling and identification via bottom-up proteomics. | Thermo Fisher (51111), Promega (V5280) |
Preparing for and Succeeding in Pre-Approval Inspections (PAI)
Within the broader thesis on FDA-industry consultation for nanotechnology product development research, Pre-Approval Inspections (PAI) represent a critical juncture. For novel nanotherapeutics, the PAI evaluates not only compliance with Current Good Manufacturing Practices (CGMP) but also the adequacy of the development and validation data supporting the product's quality, safety, and efficacy. This document provides detailed application notes and protocols to navigate the unique challenges of a PAI for a nanotechnology-based drug product.
Key PAI Focus Areas for Nanotechnology Products
PAI readiness requires demonstrating control over product development and manufacturing. For nanotech products, this involves specialized emphasis on the following areas:
Table 1: Critical PAI Focus Areas and Nanotechnology-Specific Considerations
| Focus Area | General PAI Objective | Nanotechnology-Specific Considerations & Data Requirements |
|---|---|---|
| Process Validation | Demonstrate the manufacturing process consistently produces product meeting its quality attributes. | Data linking nanoparticle synthesis parameters (e.g., energy input, mixing rates, solvent ratios) to Critical Quality Attributes (CQAs) like particle size (PDI), zeta potential, drug loading, and release profile. |
| Analytical Method Validation | Show methods are suitable for detecting and quantifying product attributes and impurities. | Validation of methods for characterizing complex nanostructures (e.g., HPLC for free vs. bound drug, DLS/SEC for aggregation, TEM for morphology). Forced degradation studies to understand nanoparticle stability. |
| Raw Materials & Controls | Ensure identity, purity, and quality of components. | Characterization of novel functional excipients (e.g., PEG-lipids, targeting ligands). Control strategies for residual solvents/catalysts from synthesis. Supplier audits for specialty materials. |
| Stability Data | Establish retest/expiry dates and storage conditions. | Real-time stability data tracking nanoparticle-specific CQAs. Evidence of physical stability (no aggregation, precipitation) and chemical stability (drug leakage, excipient degradation). |
| Facility & Equipment | Verify suitability and cleanliness to prevent contamination/mix-ups. | Dedicated equipment for nanomaterial handling where appropriate. Containment strategies for airborne nanoparticles. Cleaning validation considering nanomaterial adhesion. |
| Data Integrity | Ensure all data is ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate, +). | Audit trails for electronic systems controlling critical process parameters (e.g., in-line particle sizing). Raw data from advanced characterization instruments (e.g., spectra from DSC, XRD). |
Detailed Experimental Protocols for PAI Readiness
Protocol 1: Systematic Risk Assessment for Nanomaterial Critical Quality Attributes (CQAs)
Objective: To identify and rank potential failure modes in nanoparticle manufacturing impacting CQAs, forming the basis for the control strategy. Methodology:
- Form a Cross-Functional Team: Include members from R&D, Analytical, Manufacturing, and Quality Assurance.
- Define the Process Flow: Create a detailed process map from raw material dispensing to final drug product filling.
- Identify Potential Failure Modes: For each process step, brainstorm what could go wrong (e.g., incomplete lipid dissolution, sonication energy drift, pH adjustment error).
- Assess Risk: For each failure mode, evaluate:
- Severity (S): Impact on patient safety/efficacy (1=Low, 5=High).
- Occurrence (O): Likelihood of occurrence (1=Rare, 5=Frequent).
- Detection (D): Ability to detect failure before release (1=Easy, 5=Hard).
- Calculate Risk Priority Number (RPN): RPN = S x O x D.
- Prioritize & Plan Controls: Focus mitigation efforts (process controls, in-process tests) on failure modes with the highest RPN scores.
Table 2: Example Risk Assessment for Liposome Formation Step
| Process Step | Failure Mode | Potential Effect on CQA | S | O | D | RPN | Recommended Control Action |
|---|---|---|---|---|---|---|---|
| Sonication for Size Reduction | Inconsistent sonication time/energy | High PDI, batch non-uniformity | 4 | 3 | 2 | 24 | Automated timer & energy monitor; in-process DLS sample. |
| Tangential Flow Filtration (TFF) | Membrane fouling or breach | Change in particle size, sterility breach | 5 | 2 | 3 | 30 | Pre-use integrity testing; post-TFF size and sterility testing. |
Protocol 2: Forced Degradation Study for a Polymeric Nanoparticle Formulation
Objective: To proactively identify potential degradation pathways and impurities, and to demonstrate the stability-indicating capability of analytical methods for the PAI. Materials: Purified nanoparticle drug product batch. Stress Conditions:
- Acidic Hydrolysis: Incubate in pH 3.0 buffer at 25°C for 24 hours.
- Basic Hydrolysis: Incubate in pH 10.0 buffer at 25°C for 24 hours.
- Oxidative Stress: Incubate with 0.3% hydrogen peroxide at 25°C for 24 hours.
- Thermal Stress: Solid state and in solution at 40°C and 60°C for 1 month.
- Photostress: Expose to ICH Q1B Option 2 conditions (UV and Vis light). Analysis: After stress, apply a panel of methods to detect changes:
- HPLC/DAD: Assess drug substance degradation, new impurity peaks.
- Size Exclusion Chromatography (SEC) with MALS: Detect polymer cleavage or nanoparticle aggregation.
- Dynamic Light Scattering (DLS): Monitor changes in hydrodynamic diameter and PDI.
- Zeta Potential: Evaluate surface charge changes indicating stability shifts. Deliverable: A report linking stress conditions to specific degradation products and physical changes, proving methods can distinguish intact product from degradants.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Nanotechnology Product Development & PAI Readiness
| Item | Function & Relevance to PAI |
|---|---|
| Certified Reference Standards | Essential for analytical method validation. For nanotech, may include monodisperse particle size standards (e.g., NIST-traceable latex beads) and certified impurity standards. |
| Functionalized Lipids/Polymers | High-purity, well-characterized building blocks (e.g., DSPC, mPEG2000-DSPE, PLGA). Certificates of Analysis (CoA) with detailed impurity profiles are critical for regulatory filings. |
| In-line/On-line Particle Analyzers | Probes for real-time monitoring of particle size (e.g., FBRM, DLS) during manufacturing. Data supports process validation and demonstrates consistent control. |
| Stable Isotope-Labeled Compounds | Used as internal standards in mass spectrometry methods for quantifying drug loading or detecting excipient degradation, enhancing method robustness. |
| Advanced Microscopy Grids | Specialized grids (e.g., carbon-coated TEM grids) for high-resolution imaging (TEM, AFM) to provide visual evidence of nanoparticle morphology and uniformity. |
Visualization: PAI Readiness Workflow & Nanomaterial Characterization Pathways
Conclusion
Successful navigation of FDA consultations for nanotechnology products requires a proactive, science-driven, and holistic approach that integrates regulatory strategy with robust technical development from the earliest stages. Key takeaways include the necessity of early and iterative engagement with the FDA, the paramount importance of thorough and methodical physicochemical and biological characterization, and the strategic value of a Quality by Design framework. Future directions will involve adapting to evolving guidelines for complex generics (peptides, liposomes), leveraging advanced characterization tools (AI/ML, real-time analytics), and addressing novel challenges in cell and gene therapy nanocarriers. By mastering these elements, development teams can de-risk their programs, accelerate timelines, and ultimately deliver innovative nanomedicines that meet the FDA's stringent standards for safety, efficacy, and quality.