Nanoparticle Targeting Efficiency in Cancer Therapy: Mechanisms, Optimization, and Clinical Translation
This article provides a comprehensive analysis of nanoparticle targeting strategies for cancer therapy, tailored for researchers and drug development professionals.
Nanoparticle Targeting Efficiency in Cancer Therapy: Mechanisms, Optimization, and Clinical Translation
Abstract
This article provides a comprehensive analysis of nanoparticle targeting strategies for cancer therapy, tailored for researchers and drug development professionals. It explores the foundational principles of passive and active targeting, including the Enhanced Permeability and Retention (EPR) effect and ligand-receptor interactions. The review delves into advanced methodological approaches such as stimuli-responsive nanoplatforms and receptor-mediated targeting, while also addressing critical challenges like multidrug resistance, the protein corona effect, and physiological barriers. A comparative evaluation of targeting efficacy, supported by in vitro and in vivo data, is presented to validate different strategies. The synthesis of current knowledge aims to guide the rational design of next-generation nanotherapeutics for improved clinical outcomes.
Fundamental Principles of Nanoparticle Tumor Targeting
The Enhanced Permeability and Retention (EPR) effect represents a foundational principle in cancer nanomedicine, first described by Matsumura and Maeda in 1986 [1]. This pathophysiological phenomenon enables the passive targeting of macromolecular drugs and nanocarriers to solid tumors, forming the basis for numerous therapeutic strategies in oncology [2] [1]. The EPR effect exploits the unique anatomical and physiological abnormalities of tumor vasculature, characterized by leaky blood vessels with enlarged inter-endothelial gaps (100-780 nm) and impaired lymphatic drainage systems [2] [3]. These pathological features allow nanoscale particles (typically 10-100 nm) to extravasate from the bloodstream into tumor tissue while promoting their prolonged retention within the tumor interstitium [4] [1].
The clinical significance of the EPR effect is substantial, underpinning at least 15 clinically approved nanomedicines including Doxil and Apealea [1]. By leveraging this passive targeting mechanism, nanocarriers can achieve higher drug concentrations at tumor sites while reducing systemic exposure, potentially minimizing off-target toxicity and improving therapeutic indices [4] [3]. However, the heterogeneity of the EPR effect across different tumor types, disease stages, and individual patients presents significant challenges for clinical translation, prompting researchers to develop innovative strategies to enhance and complement this natural targeting mechanism [5] [2] [1].
Pathophysiological Basis of the EPR Effect
Tumor Vasculature Abnormalities
The structural foundation of the EPR effect lies in the aberrant vasculature of solid tumors. Unlike normal tissues with organized angiogenesis, tumors exhibit rapid, dysregulated blood vessel formation driven by hypoxia and cancer cell-derived signals [2]. This results in structurally and functionally compromised vasculature with discontinuous endothelium, incomplete basement membranes, and abnormal pericyte coverage [1]. The endothelial gaps in tumor vessels typically range from 100-780 nm, significantly larger than the 5-10 nm fenestrations found in normal continuous capillaries, thereby facilitating the extravasation of nanomedicines [2] [3].
Several molecular mediators contribute to this hyperpermeability, including vascular endothelial growth factor (VEGF), prostaglandins, bradykinin, nitric oxide, and various proteases [2]. These factors not only promote angiogenesis but also actively increase vascular permeability by inducing endothelial cell contraction and disrupting cell-cell junctions [5]. The resulting "leaky" vasculature enables the passive accumulation of nanoparticles and macromolecular therapeutics in tumor tissues, representing the "enhanced permeability" component of the EPR effect [1].
Impaired Lymphatic Drainage and Retention Mechanisms
The "retention" component of the EPR effect stems from deficient lymphatic function within tumor tissues [1]. Solid tumors typically exhibit compressed or non-functional lymphatic vessels due to rapid cancer cell proliferation and increased interstitial pressure [2] [3]. This impaired lymphatic clearance, combined with the characteristic dense extracellular matrix (ECM) of tumors, prevents the efficient removal of extravasated macromolecules and nanoparticles [2] [1].
The tumor microenvironment further promotes retention through multiple mechanisms. High collagen content, elevated hyaluronan levels, and increased stromal cell density create a physical barrier that traps nanocarriers within the tumor interstitium [2]. Additionally, elevated interstitial fluid pressure (IFP) within tumor cores, resulting from vascular leakage and poor lymphatic drainage, can approach that of the microvasculature, potentially hindering convective transport while still permitting diffusion-based nanoparticle distribution [2] [3]. These combined factors enable the prolonged retention of nanomedicines, potentially increasing their therapeutic efficacy against cancer cells [1].
Table 1: Key Pathophysiological Features Underlying the EPR Effect
| Feature | Description | Impact on EPR |
|---|---|---|
| Vascular Hyperpermeability | Discontinuous endothelium with 100-780 nm gaps | Enables extravasation of nanoparticles and macromolecules |
| Aberrant Angiogenesis | Rapid, disorganized blood vessel formation | Creates structurally abnormal vasculature prone to leakage |
| Vascular Mediators | VEGF, prostaglandins, bradykinin, nitric oxide | Actively increase vascular permeability through endothelial cell contraction |
| Deficient Lymphatic System | Compressed or non-functional lymphatic vessels | Reduces clearance of extravasated particles |
| Dense Extracellular Matrix | High collagen, hyaluronan, and stromal cell density | Traps nanoparticles within tumor interstitium |
| Elevated Interstitial Fluid Pressure | Increased pressure from vascular leakage and poor drainage | May hinder convective transport but permits diffusion |
Critical Analysis of EPR Limitations and Heterogeneity
Inter- and Intra-tumor Heterogeneity
The EPR effect demonstrates significant variability that substantially impacts its therapeutic reliability [2] [1]. This heterogeneity manifests at multiple levels, creating substantial challenges for clinical translation of EPR-dependent nanomedicines. Different tumor types exhibit markedly different EPR characteristics, with pancreatic cancers typically demonstrating less leaky vasculature compared to other solid tumors [6]. Furthermore, heterogeneity exists within individual tumors, where well-perfused peripheral regions may show robust EPR effects while hypoxic central regions exhibit limited nanoparticle accumulation due to compressed vasculature and elevated interstitial fluid pressure [2] [3].
Temporal evolution of the EPR effect presents additional complications, as early-stage tumors often possess more organized vasculature with less pronounced EPR characteristics compared to advanced, late-stage tumors [6]. This temporal variability is further complicated by species-dependent differences, with the EPR effect being more consistently observed in murine tumor models than in human patients, partially explaining the frequent discrepancy between preclinical success and clinical outcomes [6] [1]. These multifaceted heterogeneity issues necessitate careful patient stratification and the development of complementary strategies to enhance EPR-mediated drug delivery [5] [1].
Physiological and Biological Barriers
Several physiological barriers within the tumor microenvironment limit the effectiveness of the EPR effect. Elevated interstitial fluid pressure (IFP) resulting from vascular leakage and impaired lymphatic function can create pressure gradients that hinder nanoparticle penetration from vessels into the tumor interstitium [2] [3]. This elevated IFP is particularly pronounced in larger tumors, potentially explaining the limited penetration of nanomedicines into tumor cores [3].
The dense extracellular matrix (ECM) of tumors, characterized by high collagen content, increased stromal cellularity, and abnormal ECM cross-linking, creates significant physical barriers to nanoparticle distribution [2]. This dense stroma not limits diffusion but also sequesters various growth factors and cytokines that further modulate tumor pathophysiology [2] [1]. Additionally, cellular components including tumor-associated macrophages (TAMs) and other phagocytic cells can actively take up and sequester nanoparticles, potentially diverting them from their intended targets [5] [7]. These biological barriers collectively contribute to the limited penetration and heterogeneous distribution of nanomedicines within tumors, even when extravasation occurs efficiently [2] [1].
Diagram 1: Key Limitations Affecting EPR Efficacy
Quantitative Assessment of EPR Performance
Nanoparticle Delivery Efficiency Metrics
The overall delivery efficiency of nanoparticles to tumors via the EPR effect remains surprisingly low, with quantitative studies indicating that only approximately 0.7% of the injected nanoparticle dose typically accumulates in solid tumors [8]. This low accumulation efficiency represents a significant challenge for clinical translation of EPR-based therapies. The distribution of nanoparticles within tumors follows heterogeneous patterns, with peripheral regions often receiving higher nanoparticle concentrations compared to hypoxic central regions where therapeutic need may be greatest [2] [3].
Multiple factors influence this delivery efficiency, including nanoparticle physicochemical properties, tumor type, vascular density, and specific pathophysiological characteristics of individual tumors [2] [8]. The mononuclear phagocyte system (MPS), particularly macrophages in the liver and spleen, represents the primary clearance pathway for systemically administered nanoparticles, significantly reducing their circulation half-life and availability for tumor accumulation [8]. Renal clearance further depletes smaller nanoparticles (<10 nm), while opsonization and immune recognition accelerate removal from circulation, collectively contributing to the modest tumor accumulation percentages observed across numerous studies [4] [8].
Impact of Nanoparticle Design Parameters
Nanoparticle physicochemical properties significantly influence their performance in EPR-mediated tumor targeting. Size represents a critical parameter, with optimal nanoparticle diameters typically falling between 10-100 nm [4]. Particles smaller than 10 nm undergo rapid renal clearance, while those exceeding 100 nm experience increased recognition and clearance by the mononuclear phagocyte system [4] [8]. Surface characteristics, particularly hydrophilicity and charge, substantially impact circulation half-life, with PEGylation (polyethylene glycol coating) demonstrating proven benefits in reducing opsonization and extending circulation time [4] [8].
Shape and rigidity represent additional design considerations that influence margination, vascular transport, and extravasation potential [8]. The nanoparticle material composition (lipidic, polymeric, or inorganic) further affects drug loading capacity, release kinetics, and biocompatibility [9] [4]. These design parameters must be carefully optimized to maximize EPR-mediated tumor accumulation while minimizing off-target distribution and clearance [4] [8].
Table 2: Quantitative Analysis of Nanoparticle Parameters Influencing EPR Efficacy
| Parameter | Optimal Range | Impact on EPR Efficiency | Experimental Evidence |
|---|---|---|---|
| Size | 10-100 nm | <10 nm: Renal clearance>100 nm: MPS clearance | Preclinical models show 20-100 nm particles have longest circulation and highest tumor accumulation [4] [8] |
| Surface Charge | Neutral to slightly negative | Positive charge: Rapid clearanceNegative charge: Prolonged circulation | Neutral/slightly negative particles demonstrate 2-3× longer half-life than highly charged particles [4] [7] |
| Surface Modification | PEGylation | Reduces opsonization, extends circulation | PEGylated liposomes show 40-60% longer circulation half-life vs. non-PEGylated [4] [8] |
| Drug Loading Capacity | >5% w/w | Higher payload improves therapeutic efficacy | Polymeric NPs achieve 5-20% loading; dendrimers can reach 30%+ [9] [4] |
| Tumor Accumulation | ~0.7% ID/g | Only fraction reaches tumor tissue | Quantitative biodistribution studies across multiple nanoformulations [8] |
Strategies to Enhance EPR-Mediated Drug Delivery
Pharmacological and Physical Priming Approaches
Several strategies have been developed to enhance the EPR effect through pharmacological or physical modulation of the tumor microenvironment. Pharmacological approaches include using angiotensin-converting enzyme (ACE) inhibitors to normalize blood pressure and improve tumor perfusion, or administering vascular mediators such as nitric oxide donors, prostaglandins, or bradykinin analogs to actively increase vascular permeability [5] [2]. These pharmacological primers can be administered prior to nanomedicine treatment to create a more favorable environment for nanoparticle extravasation and distribution [5].
Physical priming methods utilize external energy sources to locally enhance vascular permeability and nanoparticle delivery. These include mild hyperthermia, which can expand endothelial gaps and improve nanoparticle extravasation [6]; ultrasound, particularly in combination with microbubbles (sonoporation) that mechanically disrupt vessel walls [5]; and radiation therapy, which can modulate vascular function and increase permeability in irradiated fields [5]. These physical approaches offer spatial and temporal control over EPR enhancement, potentially minimizing systemic effects while improving local drug delivery [5] [6].
Advanced Nanocarrier Design Strategies
Sophisticated nanocarrier engineering represents another promising approach to enhance EPR-mediated drug delivery. Multi-stage delivery systems incorporate size-shrinking capabilities or charge-reversal mechanisms that optimize different aspects of the delivery process [2] [1]. For example, larger carriers (100-200 nm) may demonstrate optimal vascular transport and initial extravasation, then release smaller nanoparticles (10-20 nm) that penetrate deeper into tumor tissue [2].
Stimuli-responsive nanocarriers that react to tumor-specific signals (pH, enzymes, redox status) or external triggers (light, magnetic fields) offer additional targeting precision [9] [1]. These "smart" nanoparticles can maintain stable circulation while activating their targeting or release mechanisms specifically within the tumor microenvironment [9]. Additionally, biomimetic approaches utilizing cell membrane coatings from platelets, leukocytes, or cancer cells themselves can exploit natural homing mechanisms to improve tumor accumulation while evading immune recognition [1]. These advanced design strategies aim to overcome biological barriers while leveraging the fundamental EPR effect for improved therapeutic outcomes [9] [1].
Diagram 2: EPR Enhancement Strategies
Experimental Models and Methodologies
In Vivo and Intravital Imaging Techniques
The evaluation of EPR effect efficiency relies heavily on advanced imaging methodologies that enable real-time visualization of nanoparticle distribution and tumor accumulation. Intravital microscopy (IVM) represents a powerful technique for directly observing nanoparticle extravasation, distribution, and retention in living animal models [10]. This approach provides dynamic, high-resolution information about the spatial and temporal heterogeneity of nanoparticle delivery, allowing researchers to identify specific vascular and stromal barriers that limit treatment efficacy [10].
Complementary techniques include fluorescence lifetime imaging microscopy (FLIM) and Förster resonance energy transfer (FRET), which can provide information about nanoparticle integrity and drug release kinetics within tumor tissues [10]. These advanced imaging methods have revealed critical insights into EPR heterogeneity, demonstrating how physiological factors including vascular density, perfusion efficiency, and interstitial pressure collectively influence nanoparticle distribution patterns [10] [8]. The experimental data generated through these techniques provides essential validation for EPR-based targeting strategies and informs the rational design of improved nanocarriers [10].
Standardized Evaluation Protocols
Robust assessment of EPR-mediated drug delivery requires standardized protocols for quantifying tumor accumulation and distribution. A typical experimental workflow begins with nanoparticle characterization to determine size distribution, surface charge, drug loading efficiency, and stability in physiological conditions [7]. Following intravenous administration in tumor-bearing models, researchers collect time-point samples to determine pharmacokinetic parameters including circulation half-life, volume of distribution, and clearance rates [4] [7].
Biodistribution studies quantify nanoparticle accumulation in tumors versus major organs (liver, spleen, kidneys, heart, lungs) using techniques including fluorescence imaging, radiolabeling, or mass spectrometry [7]. These studies typically express results as percentage injected dose per gram of tissue (% ID/g), allowing direct comparison between different nanocarrier formulations [8] [7]. Additional analyses may include histological assessment of nanoparticle distribution within tumor sections, penetration depth measurements, and correlation with therapeutic efficacy endpoints [10] [7]. These standardized protocols enable meaningful comparisons between different EPR-enhancement strategies and facilitate translation from preclinical models to clinical applications [7].
Research Reagent Solutions for EPR Studies
Table 3: Essential Research Reagents for EPR Effect Investigation
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Nanocarrier Platforms | PEGylated liposomes, PLGA nanoparticles, Gold nanorods, Dendrimers | Fundamental EPR effect studies; drug delivery optimization | Size control, surface modification capacity, drug loading efficiency [9] [4] |
| Imaging Agents | Fluorescent dyes (DiR, Cy5.5), Radiolabels (⁹⁹ᵐTc, ⁶⁴Cu), Contrast agents (IONPs) | Biodistribution studies; pharmacokinetic analysis; tumor accumulation quantification | Signal sensitivity, stability, compatibility with detection systems [10] [7] |
| Vascular Permeability Modulators | Nitric oxide donors, VEGF, Bradykinin analogs, ACE inhibitors | EPR enhancement studies; pharmacological priming approaches | Dosing optimization, temporal control, specificity for tumor vasculature [5] [2] |
| Tumor Model Systems | Subcutaneous xenografts, Orthotopic models, Genetically engineered models, Patient-derived xenografts | EPR heterogeneity assessment; translational relevance | Vascular characteristics, stromal composition, clinical relevance [10] [8] |
| Analytical Tools | HPLC/MS systems, Gamma counters, Fluorescence imaging systems, Histopathology platforms | Quantitative biodistribution; spatial distribution analysis | Sensitivity, quantification accuracy, spatial resolution [10] [7] |
The EPR effect remains a fundamental principle in cancer nanomedicine, providing the physiological basis for passive tumor targeting of nanotherapeutic agents. While substantial evidence supports its existence and utility, significant challenges persist due to the heterogeneity of the effect across different tumors and individual patients [5] [1]. The future of EPR-based drug delivery lies in developing personalized approaches that account for this heterogeneity through patient stratification and treatment customization [5] [1].
Emerging strategies focus on combining EPR-mediated passive targeting with active targeting mechanisms, physical enhancement methods, and sophisticated nanocarrier designs that respond to specific tumor microenvironment cues [5] [9] [1]. The integration of artificial intelligence and computational modeling approaches shows particular promise for optimizing nanoparticle design parameters and predicting EPR efficiency in individual patients [8] [1]. Additionally, advanced imaging techniques and biomarker development may enable better patient selection for EPR-based therapies, potentially identifying those most likely to benefit from nanomedicine approaches [5] [10].
As the field progresses, the successful clinical translation of EPR-based nanomedicines will likely require multimodal strategies that address the complex biological barriers of solid tumors while leveraging the fundamental principles of enhanced permeability and retention [2] [1]. Through continued refinement of nanocarrier design, enhancement techniques, and patient selection methods, the EPR effect will remain a cornerstone of targeted cancer therapy, enabling more effective and less toxic treatment options for cancer patients [5] [1].
Active targeting represents a sophisticated strategy in nanomedicine designed to enhance the specificity of therapeutic agents for diseased cells, thereby maximizing therapeutic efficacy while minimizing off-target effects. This mechanism relies on the deliberate engineering of nanocarriers with surface-bound ligands that recognize and bind to specific receptors overexpressed on target cells [3]. Contrary to the common misconception that these ligands confer a "homing" ability, active targeting functions not by attracting nanoparticles from a distance but by significantly improving their retention and uptake at the target site following passive accumulation, a process fundamentally governed by short-range chemical forces [11]. The success of this approach is a delicate balance of multiple factors, including target receptor accessibility, ligand-receptor binding kinetics, and the physicochemical properties of the nanoparticle itself [11]. This guide provides a comparative analysis of active targeting strategies, focusing on the core principles of ligand-receptor interactions, the quantitative aspects governing these interactions, and the experimental methodologies used to evaluate their efficiency in the context of cancer therapy.
Fundamental Principles of Ligand-Receptor Interactions
The efficacy of actively targeted nanoparticles is not a simple function of ligand presence; it is governed by a two-step process that requires both successful contact and subsequent molecular engagement.
A Two-Step Mechanism: Contact and Retention
The process of active targeting can be broken down into two sequential, critical steps:
- Step 1: Contact: The nanoparticle must first come into close physical proximity with the target cell. The accessibility of the target cell is a primary determinant of success. For example, vascular endothelial cells are highly accessible to intravenously administered nanoparticles, whereas reaching extravascular tumor cells requires first crossing the vascular endothelium, a significant barrier that often limits delivery to a small fraction (~1%) of the injected dose [11].
- Step 2: Retention and Capture: Once contact is made, the conjugated ligands on the nanoparticle surface can engage with their cognate cell-surface receptors. This specific binding event facilitates the retention of the nanoparticle at the target site, increasing the likelihood of cellular internalization and thus, drug delivery [11]. This step is not a long-range "magnetic" pull but a short-range interaction mediated by hydrogen bonds and electrostatic forces that act over distances of merely 0.3–0.5 nm [11].
The Critical Role of Ligand Density and Avidity
The number of ligands on a nanoparticle's surface—its ligand density—is a crucial design parameter that directly impacts avidity (the cumulative strength of multiple simultaneous interactions) and cellular selectivity. Interestingly, "more" does not always mean "better." While engineered nanoparticles are typically coated with a high density of ligands, nature provides an optimized model in viruses, which achieve high target cell avidity and selectivity with a relatively low number of receptor-binding spikes [12].
Experimental studies using polymeric nanoparticles functionalized with an angiotensin II receptor antagonist revealed that only a fraction of the surface ligands (approximately 18%) are actually involved in specific binding to target cells [12]. This suggests that a strategic optimization of ligand number, rather than simple maximization, is key to developing nanoparticles with high efficiency and specificity, mirroring the evolutionary refinement seen in viruses [12].
Quantitative Comparison of Targeting Ligands and Strategies
The selection of a targeting ligand is a fundamental decision that influences the specificity, efficiency, and overall success of a nanocarrier system. The table below provides a comparative overview of commonly used ligand classes.
Table 1: Comparison of Common Targeting Ligands Used in Active Targeting
| Ligand Class | Example Target Receptor(s) | Key Advantages | Key Limitations |
|---|---|---|---|
| Antibodies/Aptamers | Various tumor-associated antigens | High specificity and binding affinity [3] | Large size may hinder penetration; potential immunogenicity [13] |
| Peptides | Integrins (e.g., αvβ3), Insulin Receptor | Good penetration; can be designed for allosteric sites [13] | Susceptible to proteolytic degradation; moderate affinity |
| Small Molecules | Folate receptor, Carbohydrate receptors | Low immunogenicity; favorable pharmacokinetics [3] | Limited number of well-characterized targets |
| Allosteric Peptides | Transmembrane Domains (TMDs) of receptors (e.g., IR) | Avoids competition from endogenous ligands; overcomes target loss from extracellular domain shedding [13] | Novel approach; requires extensive validation |
Emerging strategies are addressing the limitations of traditional orthosteric targeting. For instance, allosteric targeted drug delivery utilizes peptide ligands designed to bind specifically to the transmembrane domains (TMDs) of receptors, such as the insulin receptor (IR) or integrin αvβ3, rather than their extracellular domains [13]. This approach offers distinct advantages:
- Avoids Competitive Interference: It is not competitively inhibited by endogenous ligands like insulin, a common issue for ligands targeting the extracellular domain of the IR [13].
- Overcomes Target Loss: It remains effective even when the extracellular domain of a receptor is shed or mutated, a phenomenon that can render conventional targeting moieties ineffective [13].
- Plug-and-Play Simplicity: These lipophilic peptides can be spontaneously embedded into the phospholipid layers of lipid-based carriers (e.g., liposomes, lipid nanoparticles) without complex chemical conjugation [13].
Table 2: Quantitative Analysis of Ligand-Receptor Binding Parameters
| Targeting Strategy | Ligand/Receptor Pair | Measured Binding Parameter | Value | Experimental Method |
|---|---|---|---|---|
| Orthosteric Targeting | Losartan (EXP3174) / AT1R | Optimal Ligand Density | 29 ligands/100 nm² (total); 5.3 ligands/100 nm² (binding) [12] | ICP-MS, Flow Cytometry |
| Allosteric Targeting | ITP Peptide / IR TMD | Dissociation Constant (KD) | 2.10 × 10⁻⁷ M [13] | Surface Plasmon Resonance (SPR) |
| Allosteric Targeting | ITP Peptide / IR TMD | Predicted Binding Free Energy | -43.59 kcal/mol [13] | MM/GBSA Molecular Modeling |
Experimental Protocols for Evaluating Targeting Efficiency
Robust experimental protocols are essential for quantifying the success of an active targeting strategy. The following sections detail key methodologies cited in recent literature.
Quantifying Ligand-Receptor Interactions on Cell Surfaces
A groundbreaking experimental approach was used to determine the exact number of ligands per nanoparticle involved in binding to target cells [12].
- Nanoparticle Model: Core-shell nanoparticles composed of PLGA and a PLA-PEG block copolymer, functionalized with EXP3174 (an AT1R antagonist) at a 25% surface density.
- Key Reagent: Selenium-tagged nanoparticles. Elemental selenium was embedded in the nanoparticle core to serve as a tag for highly sensitive quantification via Inductively Coupled Plasma Mass Spectrometry (ICP-MS) [12].
- Experimental Workflow:
- Incubate nanoparticles with AT1R-expressing rat mesangial cells.
- Use EXP3174 to saturate 50% of the cell surface receptors, ensuring the applied nanoparticle concentration equals its equilibrium inhibition constant (Ki).
- Thoroughly wash cells to remove unbound nanoparticles.
- Quantify cell-associated nanoparticles by measuring the selenium signal via ICP-MS.
- Calculate the average number of binding ligands per nanoparticle using a defined formula that considers the number of target receptors and the number of nanoparticles attached to the cell membrane [12].
Validating Allosteric Targeting Mechanisms
To confirm that a novel peptide (ITP) binds to the insulin receptor's transmembrane domain in an allosteric (non-competitive) manner, researchers employed the following protocol [13]:
- Cell Model: Primary brain microvascular endothelial cells (BMECs), which naturally express the insulin receptor.
- Experimental Workflow:
- Pre-incubate BMECs with either:
- The natural ligand, insulin (which binds the extracellular orthosteric site).
- The designed ITP peptide (targeting the TMD).
- A control scrambled peptide.
- Treat the pre-incubated cells with fluorescently labelled (FITC) insulin or fluorescently labelled ITP.
- Measure cellular uptake of the fluorescent compounds using flow cytometry.
- Pre-incubate BMECs with either:
- Key Outcome: Pre-incubation with insulin competitively inhibited the uptake of FITC-insulin but did not inhibit the uptake of FITC-ITP. Conversely, pre-incubation with ITP did not inhibit FITC-insulin uptake. This mutual non-interference conclusively demonstrated that ITP and insulin bind to distinct, non-competitive sites on the IR, validating the allosteric mechanism [13].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their functions as derived from the experimental methodologies discussed in this guide.
Table 3: Research Reagent Solutions for Active Targeting Studies
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| PLGA/PLA-PEG Copolymers | Forms biodegradable, biocompatible core-shell nanoparticles that can be functionalized with ligands [12]. | Model nanoparticle system for studying ligand density [12]. |
| Selenium Tag | An elemental tag incorporated into nanoparticles for ultra-sensitive quantification via ICP-MS [12]. | Precisely measuring the number of nanoparticles bound to cells [12]. |
| Surface Plasmon Resonance (SPR) | A biosensor technique used to characterize biomolecular interactions in real-time without labels. | Measuring the dissociation constant (KD) between a peptide ligand and its target receptor [13]. |
| Allosteric Peptide Binders (e.g., ITP) | Peptides designed to bind the transmembrane domain of receptors, avoiding orthosteric competition [13]. | Enabling targeted drug delivery to the brain without interference from endogenous insulin [13]. |
| Fluorescent Probes (e.g., FITC, CY-5) | Dyes used to label ligands, drugs, or nanoparticles for visualization and quantification. | Tracking cellular uptake via flow cytometry or confocal microscopy [13]. |
Conceptual Framework and Signaling Pathways
The following diagrams illustrate the core concepts and mechanisms described in this guide.
Diagram 1: Two-Step Mechanism of Active Targeting
This diagram visualizes the fundamental process by which ligand-functionalized nanoparticles achieve target site retention.
Diagram 2: Orthosteric vs. Allosteric Targeting Strategy
This diagram contrasts the traditional orthosteric targeting approach with the novel allosteric strategy that avoids competitive inhibition.
Active targeting through ligand-receptor interactions has moved beyond the simple concept of attaching a ligand to a nanoparticle. The field is evolving towards a more nuanced and sophisticated understanding, emphasizing the quantitative optimization of ligand density, the strategic selection of highly accessible cellular targets, and the innovative development of allosteric targeting strategies that circumvent biological limitations. The experimental data and comparative analysis presented in this guide underscore that successful targeting is a kinetic competition, where enhancing the rate of accumulation and retention at the desired site is paramount. As the toolkit of ligands and nanocarriers expands, and with the aid of advanced quantitative experimental protocols, the rational design of next-generation targeted nanotherapies continues to hold immense promise for improving the precision and efficacy of cancer treatment.
In the field of cancer therapy, nanoparticle (NP)-based drug delivery systems have emerged as a promising strategy to enhance therapeutic efficacy while reducing the debilitating side effects associated with conventional chemotherapy. The effectiveness of these systems hinges on their ability to precisely accumulate at tumor sites and efficiently penetrate the complex tumor microenvironment. This targeting efficiency is not accidental but is profoundly governed by three fundamental physicochemical properties: size, surface charge, and shape. Decades of research have demonstrated that these parameters dictate NP interactions with biological systems, influencing circulation time, cellular uptake, biodistribution, and ultimately, therapeutic outcomes. Understanding the complex interplay between these properties is essential for researchers and drug development professionals aiming to design next-generation nanomedicines. This guide provides a comparative analysis of how size, surface charge, and shape influence nanoparticle targeting, supported by experimental data and detailed methodologies to inform rational NP design in cancer therapy research.
The Impact of Nanoparticle Size
Size-Dependent Cellular Uptake and Biodistribution
Nanoparticle size is a primary determinant in governing biological interactions, from systemic circulation and tissue penetration to cellular internalization. The size range of 10-200 nm is generally considered optimal for intravenous injection, as particles smaller than 10 nm are rapidly cleared by renal filtration, while those larger than 200 nm are prone to sequestration by the spleen and liver macrophages [14] [15]. The enhanced permeability and retention (EPR) effect, a cornerstone of passive tumor targeting, allows nanoparticles of approximately 10-150 nm to extravasate through the leaky vasculature of tumors and accumulate in the tumor interstitium [15]. However, a critical trade-off exists between accumulation depth and penetration within tumor tissue.
Table 1: Size-Dependent Effects on Nanoparticle Behavior
| Size Range | Key Findings on Targeting and Behavior | Experimental Model | Citation |
|---|---|---|---|
| ~100 nm | Highest cellular uptake rate and greatest distribution in liver; superior contrast enhancement in hepatic lesions. | In vivo mouse liver MRI | [14] |
| 25-50 nm | Exhibited similar accumulation and penetration in 3D tumor spheroids, outperforming larger NPs. | 3D Spheroid Models (A549 cells) | [16] |
| 100-200 nm | Limited penetration in 3D spheroids compared to smaller NPs. | 3D Spheroid Models (A549 cells) | [16] |
| < 10 nm | Effective for deep tissue penetration; 2 nm NPs accumulated more effectively than 6 nm and 15 nm particles. | 3D Spheroid Models | [16] |
| 10-150 nm | Optimal for prolonged circulation and accumulation in tumors via the EPR effect. | Review of NP delivery | [15] |
Experimental Protocols for Studying Size Effects
Synthesis of Size-Modulated Nanoparticles: A common method for generating a series of NPs with different sizes is by modulating reactant ratios. In a study on polyvinylpyrrolidone-coated iron oxide nanoparticles (PVP-IOs), the particle core size was controlled by varying the concentration of PVP to iron carbonyl (Fe(CO)₅) during thermal decomposition. Increasing the PVP concentration from 0.07 to 0.33 g/mL resulted in a decrease in core size from 65.3 nm to 7.6 nm [14]. The hydrodynamic diameter, which includes the core and the surface coating in a hydrated state, is typically measured using Dynamic Light Scattering (DLS) and is more relevant for predicting in vivo behavior.
In Vitro and In Vivo Evaluation:
- Cellular Uptake in 2D Models: Incubate different sized NPs with cancer cell monolayers (e.g., A549 lung carcinoma cells) for a set time (e.g., 3 hours). After washing away unbound NPs, the internalized NPs can be quantified via various methods, such as measuring intrinsic photoluminescence (for gold NPs) or iron content [16].
- Penetration in 3D Models: Multicellular tumor spheroids serve as a more physiologically relevant model. Spheroids are incubated with NPs for 24 hours, then washed, fixed, and analyzed using confocal fluorescence microscopy. An in-house algorithm can be used to analyze fluorescence images and quantify NP penetration depth from the spheroid periphery to its core [16].
- In Vivo Biodistribution and Imaging: NPs are administered intravenously to animal models. Their biodistribution and accumulation in organs like the liver, spleen, and tumors can be tracked over time using imaging techniques like Magnetic Resonance Imaging (MRI). For instance, the transverse relaxivity (R2) of iron oxide NPs increases with larger core sizes, providing a darker contrast in T2-weighted MRI and enabling the quantification of liver accumulation [14].
The Role of Surface Charge
Charge-Mediated Interactions and Uptake
Surface charge, typically indicated by zeta potential, critically influences a nanoparticle's interaction with plasma proteins, blood circulation time, and cellular uptake. Positively charged (cationic) NPs generally exhibit superior cellular internalization due to strong electrostatic interactions with the negatively charged glycoproteins and phospholipids on cell membranes [17]. However, this advantage comes at a cost; cationic surfaces also promote non-specific adsorption of serum proteins (opsonins), leading to rapid clearance by the mononuclear phagocyte system (MPS) and shortened blood circulation [15] [17]. Conversely, neutral or slightly negatively charged NPs demonstrate longer circulation times due to reduced protein opsonization, but their cellular uptake is often less efficient.
Table 2: Surface Charge-Dependent Effects on Nanoparticle Behavior
| Surface Charge | Key Findings on Targeting and Behavior | Experimental Model | Citation |
|---|---|---|---|
| Negative | Consistently superior accumulation and deeper penetration in 3D tumor spheroids compared to neutral and positive NPs. | 3D Spheroid Models (A549 cells) | [16] [18] |
| Positive | Enhanced cellular internalization in 2D cell cultures due to electrostatic adhesion with cell membranes. | 2D Cell Monolayers | [17] |
| Neutral/Negative | Prolonged blood circulation by resisting protein adsorption and clearance by the reticuloendothelial system (RES). | Review of NP pharmacokinetics | [15] [17] |
| Charge-Reversal | Neutral/negative in circulation (long half-life), converts to positive in acidic tumor microenvironment (enhanced uptake). | pH-responsive NP systems | [17] |
Smart Charge-Reversal Systems and Experimental Analysis
To harness the benefits of both neutral (long circulation) and positive (high uptake) charges, researchers have developed "charge-reversal" NPs. These systems are designed to be neutral or negatively charged during blood circulation but switch to a positive charge upon encountering specific stimuli in the tumor microenvironment (TME), such as its slightly acidic pH (pH 6.5-6.8) [17].
Mechanism and Materials: A common strategy involves functionalizing NP surfaces with pH-labile bonds, such as β-carboxylic amides. For example, polylysine (PLL) amided with 2,3-dimethylmaleic anhydride (DMMA) is negatively charged at physiological pH 7.4. In the acidic TME, the β-carboxylic amide bond hydrolyzes, shedding the DMMA group and regenerating the primary amine groups of PLL, which become protonated, resulting in a negative-to-positive charge reversal [17]. The kinetics of this conversion can be tuned by the chemical structure of the anhydride.
Experimental Protocol for Zeta Potential and Uptake:
- Zeta Potential Measurement: The surface charge of NPs is measured using a zeta potential analyzer. To confirm charge-reversal, NPs are incubated in buffers mimicking physiological (pH 7.4) and tumor (pH 6.5-6.8) conditions, and their zeta potential is tracked over time [17].
- Cellular Uptake Verification: The functional consequence of charge reversal is tested by incubating NPs with cells at different pH values. A significant increase in cellular internalization at acidic pH compared to neutral pH confirms the successful operation of the charge-reversal mechanism. Flow cytometry or confocal microscopy are standard techniques for this quantification [17].
Influence of Nanoparticle Shape
Shape Effects on Tumor Penetration
While less studied than size and charge, the shape of a nanoparticle is a critical factor influencing its dynamics in the bloodstream, margination towards vessel walls, and ability to penetrate dense tumor tissue. Spherical particles have been the most widely investigated due to their ease of synthesis. However, comparisons with non-spherical shapes like rods, disks, and filaments have revealed distinct advantages and disadvantages.
Key Findings: Recent systematic studies using 3D tumor spheroids have shown that spherical NPs outperform rod-shaped NPs in both tumor accumulation and penetration depth [16]. This finding challenges earlier hypotheses that suggested high-aspect-ratio particles might navigate tissue barriers more effectively. The superior performance of spheres may be attributed to more favorable Brownian motion and easier diffusion through the porous and fibrous extracellular matrix (ECM) of tumors.
Synthesis and Evaluation of Shape-Dependent Effects
Synthesis of Shape-Controlled Nanoparticles: Gold nanoparticles (AuNPs) are a popular model for shape studies due to their highly tunable morphology. Commercially available AuNPs can be acquired as spheres (various diameters) and nanorods (various aspect ratios) with controlled surface chemistry, allowing for direct comparison while isolating the effect of shape from other variables [16].
Experimental Workflow for Shape Comparison: The protocol for evaluating shape effects is similar to that for size. Spherical and rod-shaped NPs, with other properties (e.g., surface charge) kept constant, are incubated with 2D cell monolayers and 3D tumor spheroids. After incubation and washing, internalization in 2D and penetration depth in 3D are quantified using confocal microscopy and image analysis software. This direct comparison reveals that while rods might be internalized efficiently in 2D, their penetration in more realistic 3D models can be limited [16].
Integrated Design and Experimental Toolbox
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Nanoparticle Targeting Studies
| Reagent/Material | Function and Application | Example from Literature |
|---|---|---|
| Polyvinylpyrrolidone (PVP) | A biocompatible polymer coating for iron oxide NPs; improves dispersibility, stability, and blood circulation time. | PVP-IOs for liver MRI [14] |
| Carbodiimide Crosslinkers (e.g., EDC) | Enables covalent conjugation of antibodies or targeting ligands (e.g., peptides, transferrin) to NP surfaces for active targeting. | Antibody-conjugated NPs [19] |
| 2,3-Dimethylmaleic Anhydride (DMMA) | A pH-sensitive moiety used to create charge-reversal NPs; shields positive charge until reaching acidic TME. | pH-responsive PLL-DMMA [17] |
| A549 Lung Carcinoma Cells | A well-characterized cell line capable of forming compact, tumor-like spheroids for testing NP penetration in 3D models. | 3D spheroid penetration studies [16] |
| Gold Nanoparticles (AuNPs) | A highly tunable model system with intrinsic photoluminescence; ideal for isolating effects of size, shape, and charge without fluorophore labels. | Multi-parameter studies in 3D models [16] |
| Poly(D,L-lactic-co-glycolic acid) (PLGA) | A biodegradable and FDA-approved polymer for creating polymeric NPs; drug release kinetics can be tuned by molecular weight and lactide:glycolide ratio. | Polymer-based smart nanocarriers [9] |
Visualizing the Design Logic for Optimal Targeting
The following diagram synthesizes the experimental data and illustrates the logical relationship between nanoparticle properties and their resulting biological performance, guiding the design of NPs for optimal tumor targeting.
The pursuit of efficient nanoparticle targeting in cancer therapy is a multi-parameter optimization challenge. As this guide demonstrates, the physicochemical properties of size, surface charge, and shape are not independent levers but are deeply interconnected in their influence on the biological fate of NPs. The "perfect" nanoparticle does not exist; its design must be tailored to the specific therapeutic goal, whether it is broad tumor accumulation, deep tissue penetration, or efficient cellular internalization. The emerging trend is toward "smart" nanoparticles that can dynamically change their properties in response to the tumor microenvironment, such as charge-reversal systems. Furthermore, the use of advanced 3D models for preclinical testing is proving essential for obtaining predictive data for clinical translation. By systematically understanding and applying the principles outlined in this comparison guide, researchers can make informed decisions to engineer more effective, targeted nanomedicines for oncology.
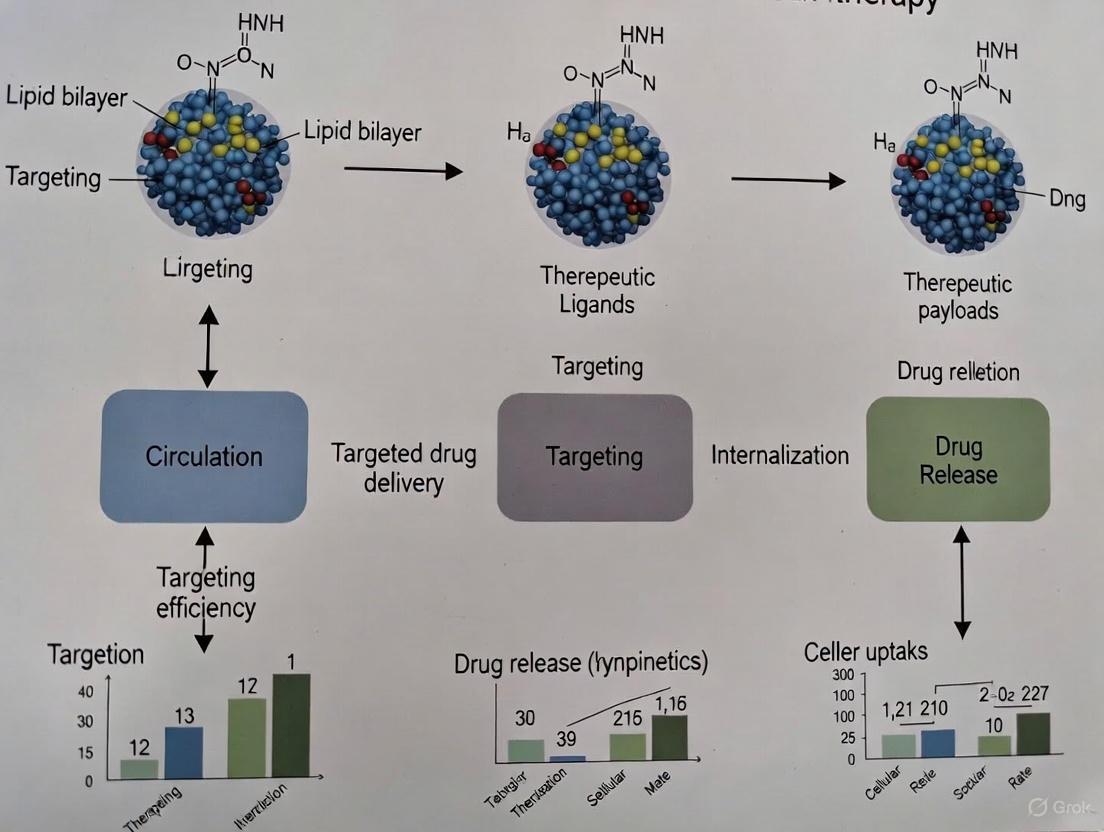
Cancer remains a leading cause of mortality worldwide, accounting for approximately 10 million deaths in 2022, with projections indicating a rise to 70 million annual deaths by 2050 [20]. Conventional chemotherapy, while effective to varying degrees, suffers from significant limitations including lack of selectivity for tumor cells, inefficient drug delivery to tumor sites, and development of multi-drug resistance [9]. The complexity of the tumor microenvironment and individual genetic variations further complicate the development of effective treatments [9]. In response to these challenges, smart nanoparticles have emerged as a transformative drug delivery platform for precise cancer therapy.
Smart nanoparticles represent an advanced class of nanocarriers engineered to respond to biological cues or be guided by them, establishing an intelligent treatment modality [9]. Unlike conventional nanoparticles, these sophisticated systems can be triggered by specific stimuli and target specific sites with precise drug delivery control [9]. After modification or activation by corresponding factors, smart nanoparticles efficiently accumulate at target locations and release their therapeutic payloads in a controlled manner [9]. Their capability to co-deliver therapeutics and diagnostic reagents has significantly advanced the development of theranostics in oncology [9]. This review provides a comprehensive classification and comparison of smart nanoparticle systems—polymeric, lipid-based, inorganic, and hybrid—within the context of their targeting efficiency in cancer therapy research.
Classification and Characterization of Smart Nanoparticles
Polymeric Nanoparticles
Polymeric nanoparticles play a pivotal role in biomedical applications, bringing together biologists, chemists, engineers, and physicians in unique collaborative ways [9]. These nanoparticles offer several advantages over non-encapsulated drugs, including improved circulation time, enhanced stability, controlled structural decomposition, higher encapsulation rates, and reduced premature and nonspecific release kinetics [9]. The development of stimuli-responsive polymeric systems represents a significant advancement in smart nanocarriers for cancer therapy.
Polymeric nanoparticles can be synthesized with combinations of both inorganic and organic components to achieve synergistic properties [9]. For instance, the combination of multiple materials can alter biological distribution, improve solubility, and enhance system stability [9]. The ability to link materials with one another can prolong blood circulation while maintaining biological effects [9]. Synthetic tunability enables the creation of smart nanoparticles that can simultaneously serve several therapeutic or imaging goals by co-encapsulating various therapeutic compounds with different release profiles [9]. A key intelligent feature of these systems is their ability to be triggered by controlling local induction of endogenous physical parameters such as electrical, thermal, ultrasound, or magnetic energy [9].
Table 1: Key Characteristics of Polymeric Nanoparticles
| Characteristic | Description | Impact on Cancer Therapy |
|---|---|---|
| Composition | Biodegradable polymers (PLGA, PLA, chitosan, alginate) [20] | Improved biocompatibility and reduced toxicity compared to non-biodegradable polymers |
| Drug Release Kinetics | Adjustable by molecular weight, lactide to ethyl ester ratio, and drug concentration [9] | Enables controlled release profiles tailored to specific cancer types |
| Surface Modification | PEGylation for stealth properties; ligand attachment for active targeting [9] | Prolonged circulation time and enhanced tumor-specific accumulation |
| Stimuli-Responsiveness | Response to pH, temperature, enzymes, or external triggers [9] | Precise drug release within tumor microenvironment |
| Manufacturing | Emulsion polymerization, solvent evaporation, salting-out, dialysis [9] | Scalable production with good repeatability |
Lipid-Based Nanocarriers
Lipid-based nanocarriers (LNs) represent one of the most established categories of nanocarriers with excellent biocompatibility profiles [21]. These systems are generally non-spherical in shape, determined by electrostatic interactions between polar/ionogenic phospholipid heads and the solvent, as well as non-polar lipid hydrocarbon moieties present in the solvent [21]. The unique physicochemical properties of lipid-based systems in the form of liposomes or solid core lipid nanoparticles make them outstanding candidates as carriers for drug delivery applications [21].
Liposomes
Liposomes, first developed in the 1960s, are composed of phospholipid bilayers similar to plasma membranes of human cells, granting them exceptional biocompatibility and the ability to promote drug diffusion across plasma membranes [21]. These self-assembled vesicles comprise one or multiple concentric lipid bilayers that enclose an aqueous core, typically ranging from 20 nm to over 1 μm in size [21]. This distinctive structure enables liposomes to hold and stabilize hydrophilic drugs in the aqueous core while encapsulating lipophilic drugs within the lipid bilayer, contributing to their remarkable versatility [21]. The landmark approval of Doxil by the FDA in 1995 as the first long-circulating liposome for cancer treatment paved the way for numerous liposomal formulations [21] [20]. When compared to free doxorubicin, Doxil significantly reduced cardiotoxicity and myelotoxicity while achieving higher drug concentrations in tumors [20].
Solid-Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs)
Solid-lipid nanoparticles (SLNs), developed in the 1990s, combine the advantages of polymer nanocarriers (strong drug loading capacity, controllable drug delivery) with the excellent biocompatibility of lipid emulsions [21]. The main feature of SLNs is that they contain lipids that remain solid at room temperature, typically composed of biocompatible substances such as triglycerides, fatty acids, steroids, and biowaxes [21]. Due to their small sizes and large surface area, SLNs are suitable for surface functionalization with ligands, antibodies, and other functional groups [21]. SLNs can be orally administered as aqueous dispersions or in dosage forms such as capsules, tablets, and pellets, positioning them at the forefront of potential applications in oral drug delivery systems [21].
Nanostructured lipid carriers (NLCs) were developed as an enhancement to SLNs by replacing a fraction of solid lipids with liquid lipids to form a nanostructured matrix [21]. Unlike SLNs, the lipid matrix of NLCs consists of a mixture of solid and liquid lipids with controlled levels that possess improved capacity for bioactive retention along with controlled release attributes [21]. Since drugs generally have higher solubility in oil than in solid lipids, NLCs typically demonstrate stronger encapsulation ability for drugs compared to SLNs [21].
Table 2: Comparative Analysis of Lipid-Based Nanocarriers
| Parameter | Liposomes | Solid-Lipid Nanoparticles (SLNs) | Nanostructured Lipid Carriers (NLCs) |
|---|---|---|---|
| Structure | Phospholipid bilayer with aqueous core [21] | Solid lipid matrix at room temperature [21] | Mixture of solid and liquid lipids [21] |
| Size Range | 20 nm to >1 μm [21] | 50-1000 nm [20] | 50-1000 nm [21] |
| Drug Encapsulation | Hydrophilic (aqueous core), hydrophobic (lipid bilayer) [21] | Predominantly hydrophobic drugs [21] | Enhanced for both hydrophilic and hydrophobic [21] |
| Loading Capacity | Limited bilayer space for hydrophobic drugs [21] | Moderate [21] | High [21] |
| Key Advantages | Excellent biocompatibility, versatile drug loading [21] | Good stability, controlled release, scale-up production [21] | Improved drug loading, reduced drug expulsion [21] |
| Clinical Status | Multiple approved products (e.g., Doxil) [20] | Under investigation | Under investigation |
Inorganic Nanoparticles
Inorganic nanoparticles constitute a distinct class of nanocarriers with unique physical properties that make them particularly valuable for theranostic applications in cancer therapy. This category includes mesoporous silica nanoparticles, gold nanoparticles, iron oxide nanoparticles, quantum dots, and carbon nanotubes [9]. These materials typically exhibit characteristics such as high surface-to-volume ratio, enhanced electrical conductivity, superparamagnetic behavior, spectral shift of optical absorption, and unique fluorescence properties [21].
Gold nanoparticles possess exceptional optical properties and surface plasmon resonance that can be exploited for both imaging and photothermal therapy applications [9]. Iron oxide nanoparticles offer superparamagnetic properties suitable for magnetic resonance imaging (MRI) and magnetic hyperthermia treatments [9]. Mesoporous silica nanoparticles feature tunable pore structures that enable high drug loading capacities and surface functionalization for targeted delivery [9]. Carbon nanotubes exhibit unique electrical and thermal properties, while quantum dots provide superior fluorescence for imaging and tracking applications [9].
Hybrid Nanoparticles
Hybrid nanoparticles represent a convergence of multiple material systems designed to overcome the limitations of single-component nanocarriers. These sophisticated systems combine polymers with inorganic or organic base systems, resulting in remarkable improvements in drug targeting capabilities [22]. The development of hybrid polymer materials can circumvent the need for synthesizing entirely new molecules—an expensive process that can take several years to reach proper elaboration and approval [22].
The combination of properties in a single hybrid system confers several advantages over non-hybrid platforms, including improvements in circulation time, structural disintegration resistance, high stability, reduced premature release, enhanced encapsulation rates, and minimized unspecific release kinetics [22]. Lipid-polymer hybrid nanoparticles (LPHNs), for instance, combine the benefits of both liposomal and polymeric systems, exhibiting high drug-loading capacity, superior stability, enhanced biocompatibility, rate-limiting controlled release, prolonged drug half-lives, and improved therapeutic efficacy [20]. These hybrid systems effectively mitigate the individual disadvantages of their component materials while amplifying their beneficial characteristics [20].
Experimental Characterization Methodologies
Physicochemical Characterization Protocols
Comprehensive characterization of nanomaterial surfaces is critically important to establish structure-property relationships and provide feedback in nanomaterial design, as physiochemical characteristics directly affect performance [23]. The key parameters requiring analysis include size, shape, core structure, surface ligands, surface charge, hydrophobicity, ligand shell thickness, binding affinity, and surface morphology [23].
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy provides comprehensive structural information by analyzing the chemical environment of nuclei and has become one of the most versatile techniques for characterizing surface ligand structures [23]. In nanomaterial characterization, NMR can confirm ligand immobilization, study ligand structure, differentiate between bound and unbound ligands, quantify bound ligands, understand ligand binding mode and dynamics, and study interactions with biomolecules [23].
Experimental Protocol:
- Sample Preparation: Prepare nanoparticle dispersions in deuterated solvents at concentrations sufficient to detect surface ligands (larger nanoparticles require more sample due to dilution of surface ligands by the bulk nanomaterial) [23].
- Data Acquisition: Conduct 1H NMR experiments to confirm successful surface modification by comparing functionalized nanomaterials with free ligands [23].
- Advanced Analysis: Employ 2D-NMR techniques (DOSY, NOESY, TOCSY, HSQC, ROESY) for additional structural information [23].
- Quantitative Analysis: Use peak integration for bound ligand quantification and T2 relaxation analysis to study chain packing density and headgroup motions [23].
One significant challenge in NMR characterization of nanomaterials is line broadening of ligand signals, which becomes more severe with increasing nanoparticle size due to slower rotational correlation times [23]. Murphy and colleagues demonstrated the application of 1H NMR to characterize (11-mercaptohexadecyl)trimethylammonium bromide (MTAB) on gold nanospheres, confirming ligand attachment and studying packing density in a particle size-dependent manner [23].
Surface Charge and Hydrophobicity Analysis
Surface charge significantly influences nanoparticle behavior in biological systems, affecting cellular uptake, biodistribution, and toxicity [23]. Zeta potential measurement represents the most common method for determining nanoparticle surface charge, reflecting the electrical potential at the slipping plane of particles in solution [23].
Experimental Protocol:
- Sample Preparation: Dilute nanoparticle dispersions in appropriate buffers to maintain physiological relevance while ensuring optimal concentration for measurement.
- Instrument Calibration: Standardize zeta potential analyzer using reference materials with known zeta potential values.
- Measurement Conditions: Conduct measurements at consistent temperature (typically 25°C) with multiple runs (minimum n=3) to ensure reproducibility.
- Data Analysis: Calculate mean zeta potential values and standard deviations across replicates; interpret results in context of surface functionality.
Hydrophobicity characterization typically involves hydrophobic interaction chromatography or fluorescence-based methods using environment-sensitive probes [23]. These techniques provide insights into how nanomaterials interact with biological membranes and proteins, crucial for understanding their behavior in vivo.
Targeting Efficiency Assessment
Evaluating the targeting efficiency of smart nanoparticles requires sophisticated experimental protocols that quantify accumulation at tumor sites and specific cellular uptake.
Experimental Protocol for In Vitro Targeting Assessment:
- Cell Culture Preparation: Establish appropriate cancer cell lines (e.g., MCF-7 for breast cancer, A549 for lung cancer) and maintain under standard conditions.
- Ligand Functionalization: Conjugate targeting ligands (antibodies, peptides, aptamers, folic acid, transferrin) to nanoparticle surfaces using appropriate chemistry [9] [20].
- Competitive Binding Assays: Incubate functionalized nanoparticles with target cells in the presence and absence of free ligands to demonstrate binding specificity.
- Cellular Uptake Quantification: Employ flow cytometry or confocal microscopy with fluorescently labeled nanoparticles to quantify internalization.
- Data Analysis: Calculate targeting efficiency as the ratio of cellular uptake between targeted and non-targeted formulations.
Experimental Protocol for In Vivo Targeting Assessment:
- Animal Model Establishment: Implement appropriate tumor xenograft models (e.g., nude mice with subcutaneous or orthotopic tumors).
- Nanoparticle Administration: Administer fluorescently labeled or radiolabeled nanoparticles via intravenous injection.
- Biodistribution Analysis: At predetermined time points, euthanize animals, collect major organs and tumors, and quantify nanoparticle accumulation using imaging or analytical techniques.
- Histological Validation: Process tumor tissues for histological analysis to confirm nanoparticle localization at cellular and subcellular levels.
- Statistical Analysis: Compare tumor accumulation between targeted and non-targeted systems using appropriate statistical tests.
Diagram 1: Smart Nanoparticle Targeting Pathway in Cancer Therapy
Comparative Performance Analysis
Targeting Efficiency Across Nanoparticle Classes
The targeting efficiency of smart nanoparticles depends on multiple factors including size, surface properties, ligand density, and responsiveness to tumor microenvironment cues [9] [20]. Passive targeting leverages the enhanced permeability and retention (EPR) effect, which takes advantage of the leaky nature of tumor vasculature that allows nanoparticles to extravasate and accumulate in tumor tissue [20]. Active targeting incorporates specific ligands on nanoparticle surfaces that recognize and bind to receptors overexpressed on cancer cells [9].
Table 3: Targeting Efficiency of Smart Nanoparticle Classes
| Nanoparticle Class | Targeting Mechanisms | Ligand Functionalization | Experimental Targeting Efficiency | Key Limitations |
|---|---|---|---|---|
| Polymeric Nanoparticles | EPR, stimuli-responsive release (pH, temperature, enzymes) [9] | Antibodies, peptides, aptamers, folic acid, transferrin [9] | 3-5x higher tumor accumulation vs free drug [9] | Potential polymer toxicity, batch-to-batch variability |
| Liposomes | EPR, passive and active targeting [21] | Antibodies, peptides, carbohydrates [21] | 2-10x higher tumor concentration vs free drug (clinical data for Doxil) [21] [20] | Limited bilayer space for hydrophobic drugs, stability issues |
| Solid Lipid Nanoparticles | EPR, lymphatic targeting [21] | Proteins, surfactants, antibodies [21] | 3-8x increased bioavailability vs conventional formulations [21] | Drug expulsion during storage, low loading capacity |
| Inorganic Nanoparticles | EPR, magnetic guidance, external stimulus activation [9] | Silanes, thiols, phosphates, carboxylates [9] | 4-15x higher tumor accumulation with external magnetic field [9] | Potential long-term toxicity, slow biodegradation |
| Hybrid Nanoparticles | Combined mechanisms from component materials [22] | Multiple ligand types simultaneously [22] | 5-12x improved tumor suppression vs single-component systems [22] | Complex manufacturing, characterization challenges |
Drug Release Kinetics and Therapeutic Outcomes
Controlled drug release represents a critical parameter determining therapeutic efficacy and side effect profiles. Smart nanoparticles can be engineered to respond to various internal stimuli (pH, enzymes, redox potential) or external triggers (light, magnetic field, ultrasound) to achieve spatiotemporal control over drug release [9].
Diagram 2: Drug Release Mechanisms in Smart Nanoparticles
Experimental data from preclinical studies demonstrates that smart nanoparticles can maintain drug concentrations within the therapeutic window for extended periods, significantly improving treatment outcomes while reducing systemic toxicity. For example, DOXIL (pegylated liposomal doxorubicin) shows a significantly altered pharmacokinetic profile compared to free doxorubicin, with a half-life of approximately 55 hours versus 0.2 hours for the free drug, contributing to enhanced tumor accumulation and reduced cardiotoxicity [20].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful development and evaluation of smart nanoparticles require specialized reagents and materials tailored to specific nanoparticle classes and research objectives.
Table 4: Essential Research Reagents for Smart Nanoparticle Development
| Reagent Category | Specific Examples | Function in Nanoparticle Development | Key Considerations |
|---|---|---|---|
| Polymer Materials | PLGA, PLA, PEG, chitosan, alginate, poly(amino acids) [9] [20] | Form nanoparticle matrix, control drug release kinetics, provide biodegradability | Molecular weight, lactide:glycolide ratio (for PLGA), degree of deacetylation (for chitosan) |
| Lipid Components | Phospholipids (HSPC, DPPC), cholesterol, solid lipids (triglycerides, fatty acids) [21] | Form lipid bilayers or solid matrices, encapsulate drugs, determine stability | Phase transition temperature, purity, oxidation susceptibility |
| Surface Ligands | Antibodies, peptides (RGD), aptamers, folic acid, transferrin [9] | Enable active targeting to cancer cells, enhance cellular uptake | Binding affinity, density on nanoparticle surface, orientation |
| Characterization Reagents | NMR solvents, zeta potential standards, fluorescence dyes [23] | Facilitate physicochemical characterization and tracking | Compatibility with nanoparticle materials, stability, interference |
| Stimuli-Responsive Elements | pH-sensitive linkers, enzyme-cleavable peptides, thermosensitive polymers [9] | Enable triggered drug release in response to specific stimuli | Sensitivity, specificity, response kinetics |
The classification of smart nanoparticles into polymeric, lipid-based, inorganic, and hybrid systems provides a framework for understanding their distinct characteristics and applications in cancer therapy. Each category offers unique advantages: polymeric nanoparticles excel in controlled release and functional versatility; lipid-based systems provide exceptional biocompatibility and clinical translation potential; inorganic nanoparticles offer unique physical properties for theranostics; while hybrid systems combine beneficial properties from multiple material classes [21] [9] [22].
The future of smart nanoparticles in cancer therapy lies in advancing personalization through patient-specific design, developing multi-responsive systems that react to multiple stimuli simultaneously, and creating adaptive nanoparticles capable of modifying their properties in response to changing biological environments [9] [24]. The integration of artificial intelligence in nanoparticle design and the development of computational models to predict nanoparticle behavior in biological systems represent promising avenues to accelerate clinical translation [9]. As characterization techniques continue to improve, providing deeper insights into nanomaterial-biological interactions, the rational design of smart nanoparticles with enhanced targeting efficiency and therapeutic outcomes will undoubtedly advance, ultimately benefiting cancer patients through more effective and less toxic treatment options.
The success of nanoparticle-based cancer therapeutics relies on their efficient tumor uptake and retention [25]. The tumor microenvironment (TME) represents a complex, dynamic ecosystem that plays a pivotal role in cancer progression, therapeutic resistance, and response to treatment [26] [27] [28]. For researchers developing nanomedicines, understanding the biological characteristics of the TME is paramount, as it creates substantial barriers that significantly limit nanoparticle accumulation at the tumor site [29] [27].
The TME is composed of both cellular and non-cellular components, including cancer-associated fibroblasts (CAFs), endothelial cells, pericytes, diverse immune cells (T cells, macrophages, dendritic cells), and the extracellular matrix (ECM) [27] [30]. This microenvironment is not merely a passive bystander but actively participates in creating a hostile milieu characterized by immunosuppression, hypoxia, acidosis, and abnormal vasculature [27] [31]. These interconnected features collectively impede nanomedicine delivery through physical barriers, biochemical resistance mechanisms, and immune-mediated clearance pathways [25] [29].
Despite promising in vitro results, the clinical translation of nanoparticle formulations remains limited, with many promising preclinical studies failing to achieve expected efficacy in clinical stages [25] [26]. This translation gap highlights the critical importance of understanding TME biology and developing sophisticated nanoparticle designs that can overcome these barriers for improved therapeutic outcomes in cancer patients.
Key Biological Characteristics of the TME That Hinder Nanoparticle Accumulation
Abnormal Tumor Vasculature and Hemodynamic Barriers
The tumor vasculature system exhibits significant structural and functional abnormalities that profoundly impact nanoparticle delivery [27] [31]. Unlike normal blood vessels, tumor vessels are typically disorganized, tortuous, and highly permeable, leading to heterogeneous blood flow and oxygen distribution [27]. This dysfunctional vascular network creates substantial challenges for nanoparticle delivery:
- Heterogeneous Distribution: The enhanced permeability and retention (EPR) effect, while historically leveraged for passive targeting, results in uneven nanoparticle distribution due to variable vessel permeability and regions of hypoperfusion [29].
- Elevated Interstitial Fluid Pressure: Leaky vasculature combined with impaired lymphatic drainage creates elevated interstitial fluid pressure, reducing convective transport and limiting nanoparticle penetration into the tumor core [29] [31].
- Inadequate Perfusion: The disorganized architecture with dead ends and shunts reduces effective perfusion, creating regions that nanoparticles cannot access through circulation [27].
Table 1: Vascular Abnormalities in the TME and Impact on Nanoparticles
| Vascular Abnormality | Impact on Nanoparticle Accumulation | Potential Mitigation Strategies |
|---|---|---|
| Structural disorganization | Heterogeneous distribution | Size-tunable nanoparticles |
| Increased permeability | Primarily peripheral accumulation | Multi-stage delivery systems |
| Elevated interstitial pressure | Reduced deep penetration | Vasculature normalization agents |
| Irregular blood flow | Inconsistent delivery | Stimuli-responsive release systems |
Dense Extracellular Matrix and Physical Barriers
The extracellular matrix in solid tumors undergoes significant remodeling, creating a dense physical barrier that severely restricts nanoparticle movement [27] [32]. CAFs are primarily responsible for secreting and cross-linking ECM proteins such as collagen, fibronectin, and hyaluronic acid, dramatically increasing tissue stiffness and forming a fibrotic capsule around the tumor [27] [30]. This ECM remodeling:
- Limits Diffusion: The dense, cross-linked matrix creates narrow interstitial spaces that physically obstruct nanoparticle transport, particularly for larger formulations (>50 nm) [27].
- Traps Nanoparticles Peripherally: Nanoparticles often become trapped in the perivascular regions, unable to penetrate deeper tumor regions where the most aggressive cancer cells typically reside [27].
- Promotes Drug Resistance: The physical barrier protects interior tumor cells from therapeutic agents, contributing to treatment resistance and disease recurrence [28].
The biomechanical properties of the ECM are increasingly recognized as a major determinant of nanoparticle distribution, with studies showing that stromal targeting approaches can enhance nanomedicine penetration and efficacy [27] [32].
Immune-Mediated Clearance and Phagocytic Activity
The TME contains numerous immune cells that can recognize and clear nanoparticles, significantly reducing their circulation time and tumor accumulation [25] [30]. The mononuclear phagocyte system (MPS), particularly tumor-associated macrophages (TAMs), plays a dominant role in this clearance mechanism [25] [27]. Key aspects include:
- MPS Recognition: Opsonins in the blood serum adsorb to nanoparticles, making them recognizable to macrophages in the liver, spleen, and bone marrow, leading to rapid clearance from circulation [25].
- TAM-Mediated Uptake: Once within the TME, TAMs can internalize nanoparticles before they reach cancer cells, effectively diverting them from their intended targets [25] [30].
- Ligand-Dependent Clearance: Functionalizing nanoparticles with targeting moieties can paradoxically increase immune recognition. For instance, RGD peptide-functionalized nanoparticles demonstrated enhanced clearance by the MPS despite improved cancer cell uptake in vitro [25].
This immune-mediated clearance represents a significant challenge, particularly for actively targeted nanoparticles, and underscores the importance of evaluating targeting strategies in immunocompetent models for physiologically relevant assessments [25].
Metabolic Dysregulation: Hypoxia and Acidosis
The TME exhibits profound metabolic alterations that directly impact nanoparticle behavior and efficacy [27] [28] [31]. Rapid tumor proliferation outstrips the oxygen supply, creating hypoxic regions that trigger a shift toward anaerobic glycolysis (the Warburg effect), resulting in lactate accumulation and acidosis (pH 6.7-7.1) [27] [31]. These metabolic conditions:
- Promote Pro-Tumor Signaling: Hypoxia stabilizes hypoxia-inducible factors (HIFs) that drive the expression of pro-angiogenic factors, ECM remodeling enzymes, and immunosuppressive cytokines [27].
- Impair Immune Function: An acidic environment directly suppresses T-cell and natural killer (NK) cell function, reducing cytokine production and cytotoxic activity [30] [31].
- Alter Nanoparticle Performance: The acidic pH can affect nanoparticle stability, drug release kinetics, and surface chemistry, potentially compromising their therapeutic function [29].
Table 2: Metabolic Features of the TME Affecting Nanoparticle Performance
| Metabolic Feature | Effect on TME | Impact on Nanoparticles |
|---|---|---|
| Hypoxia | Upregulates HIF-1α, promoting angiogenesis and invasion | Can trigger release in hypoxia-responsive systems |
| Acidosis (pH 6.7-7.1) | Suppresses immune cell function, promotes invasion | Enables pH-triggered drug release in acidic compartments |
| Lactate accumulation | Creates immunosuppressive environment, drives fibrosis | Can affect surface charge and stability |
| Nutrient competition | Limits immune cell metabolism and function | May alter cellular uptake mechanisms |
Experimental Models and Methodologies for Evaluating Nanoparticle-TME Interactions
In Vitro Models: From Monolayers to 3D Systems
Advanced in vitro models provide valuable platforms for initial screening of nanoparticle interactions within TME-like conditions [25]. While traditional 2D monolayers offer simplicity and reproducibility, they lack the complexity of the native TME [25]. More physiologically relevant systems include:
- 3D Spheroid Models: These models replicate key in vivo characteristics of the TME, including concentration gradients, cellular heterogeneity, and intricate cell-cell interactions, all of which influence NP penetration and intracellular accumulation [25].
- Organoid Systems: Patient-derived organoids preserve aspects of the original TME architecture and cellular composition, serving as valuable avatars for predicting treatment response [29].
- Advanced Co-culture Systems: Incorporating multiple cell types (cancer cells, CAFs, immune cells) better mimics the cellular crosstalk within the TME and its impact on nanoparticle behavior [25].
In a comprehensive study evaluating RGD-functionalized gold nanoparticles (GNPs), both monolayer and 3D spheroid models of KPCY murine pancreatic cancer cells were utilized [25]. The researchers employed inductively coupled plasma-mass spectrometry (ICP-MS) to quantitatively measure GNP content in cells harvested at 1 h, 8 h, and 24 h timepoints, complemented by confocal imaging to confirm intracellular accumulation [25].
In Vivo Models: Importance of Immunocompetent Systems
The choice of in vivo models critically influences the assessment of nanoparticle performance, particularly regarding immune interactions [25]. While immunocompromised models (e.g., nude mice) are commonly used for their convenience, they do not accurately account for immune-related interactions, potentially leading to overestimation of targeting efficacy [25]. Key considerations include:
- Syngeneic Tumor Models: Immunocompetent models with mouse tumors derived from the same genetic background preserve intact immune systems, allowing for realistic evaluation of immune-mediated nanoparticle clearance [25].
- Biodistribution Studies: Quantitative tracking of nanoparticle distribution to tumors and major organs (liver, spleen, kidneys) provides critical data on targeting efficiency and potential off-target accumulation [25].
- Pharmacokinetic Profiling: Assessing circulation half-life, clearance rates, and temporal distribution patterns offers insights into nanoparticle behavior in physiological conditions [25].
The critical importance of model selection was demonstrated in the RGD-GNP study, where RGD functionalization significantly increased GNP uptake in cancer cells in vitro but substantially reduced tumor accumulation in vivo due to enhanced off-target clearance by the MPS, with elevated accumulation in the spleen and liver [25]. This paradoxical effect highlights how immune-driven clearance mechanisms can fundamentally alter nanoparticle performance in immunocompetent environments [25].
Diagram 1: Biological Barriers Limiting Nanoparticle Accumulation in the TME. This flowchart illustrates the major pathways that nanoparticles encounter after administration, highlighting how immune clearance, physical barriers, and metabolic conditions collectively reduce tumor accumulation. IFP = Interstitial Fluid Pressure.
Quantitative Comparison of Nanoparticle Performance in Different TME Conditions
Comparative Analysis of Targeted vs. Non-Targeted Nanoparticles
The strategic functionalization of nanoparticles with targeting moieties represents a common approach to improve tumor localization, but its efficacy must be evaluated in physiologically relevant TME conditions [25] [33]. A comprehensive study directly compared PEGylated gold nanoparticles (GNPs) with those functionalized with linear (lRGD) or cyclic (cRGD) peptides in both in vitro and in vivo settings [25]. The quantitative results reveal critical insights:
- In Vitro Performance: RGD functionalization significantly enhanced cellular uptake, with approximately 100-fold increase for GNP-PEG-lRGD and 150-fold increase for GNP-PEG-cRGD compared to non-targeted GNPs at 1-hour post-exposure [25].
- In Vivo Discrepancy: Despite superior in vitro performance, RGD-functionalized GNPs showed significantly reduced tumor accumulation in immunocompetent syngeneic mouse models, with concomitant increases in liver and spleen accumulation [25].
Table 3: Quantitative Comparison of RGD-Functionalized vs. Non-Targeted Gold Nanoparticles
| Parameter | GNP-PEG (Non-targeted) | GNP-PEG-lRGD | GNP-PEG-cRGD |
|---|---|---|---|
| In Vitro Uptake (1h) | Baseline | ~100x increase | ~150x increase |
| In Vitro Uptake (24h) | Baseline | ~20x increase | ~40x increase |
| In Vivo Tumor Accumulation | Higher | Significantly reduced | Significantly reduced |
| Liver/Spleen Accumulation | Lower | Elevated | Elevated |
| Cancer Cell Specificity | Lower | Enhanced in vitro | Enhanced in vitro |
| Immune Cell Uptake | Lower | Enhanced MPS clearance | Enhanced MPS clearance |
Methodological Framework for Evaluating Nanoparticle-TME Interactions
Robust assessment of nanoparticle behavior in the TME requires a multi-faceted methodological approach that captures both quantitative distribution and functional outcomes [25]. Key experimental protocols include:
- Nanoparticle Synthesis and Characterization: In the RGD-GNP study, researchers synthesized spherical GNPs (~12 nm core diameter) using citrate reduction methods, then functionalized them with 2 kDa thiol-terminated PEG at a grafting density of 1 PEG molecule per nm² of GNP surface area [25]. RGD peptides were added at a ratio of 1 RGD per 2 PEG molecules [25]. Characterization included transmission electron microscopy (TEM) for core size, dynamic light scattering for hydrodynamic diameter, UV-vis spectroscopy for peak absorbance, and zeta potential measurements for surface charge [25].
- Biodistribution Quantification: Following in vivo administration, ICP-MS provides highly sensitive quantification of nanoparticle accumulation in tumors and major organs, offering precise quantitative data on targeting efficiency and off-target distribution [25].
- Cellular Localization Studies: Confocal imaging and electron microscopy techniques enable visualization of nanoparticle distribution at the cellular and subcellular levels, providing spatial context to complement quantitative data [25].
- Immune Cell Interactions: Specific evaluation of nanoparticle uptake by macrophages and other immune cells through flow cytometry or specialized co-culture assays helps elucidate immune-mediated clearance pathways [25].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 4: Essential Research Tools for Studying Nanoparticle-TME Interactions
| Reagent/Methodology | Primary Function | Research Application |
|---|---|---|
| PEGylated Gold Nanoparticles | Versatile nanoplatform with tunable surface chemistry | Model system for studying TME penetration and immune interactions |
| RGD Peptides (linear/cyclic) | Targeting ligands for ανβ3 integrin receptors | Active targeting studies to evaluate specificity and clearance |
| ICP-MS | Elemental quantification with high sensitivity | Precise measurement of nanoparticle biodistribution |
| Confocal Microscopy | High-resolution 3D imaging with optical sectioning | Visualization of nanoparticle localization within tumors |
| Syngeneic Mouse Models | Immunocompetent animals with intact immune systems | Physiologically relevant assessment of immune-mediated clearance |
| 3D Spheroid Cultures | In vitro model with TME-like characteristics | Intermediate screening platform between 2D and in vivo models |
| HIF-1α Inhibitors | Modulators of hypoxic signaling | Investigating hypoxia-induced barriers to nanoparticle delivery |
| Hyaluronidase | ECM-degrading enzyme | Studying ECM remodeling to enhance nanoparticle penetration |
The biological characteristics of the tumor microenvironment present formidable yet navigable barriers to effective nanoparticle accumulation. The paradoxical findings with RGD-functionalized nanoparticles - where enhanced in vitro targeting correlated with reduced in vivo accumulation - underscore the critical importance of evaluating nanomedicines in physiologically relevant, immunocompetent models [25]. The complex interplay between abnormal vasculature, dense ECM, immune clearance mechanisms, and metabolic dysregulation requires sophisticated, multi-faceted approaches to nanoparticle design [25] [29] [27].
Future directions should focus on developing TME-modulating strategies that temporarily normalize these biological barriers rather than simply attempting to overcome them [29] [27]. This might include ECM-remodeling enzymes to reduce physical barriers, immune-modulating agents to reduce clearance, or vascular normalization approaches to improve perfusion [27]. Additionally, the development of smart nanoparticles that respond to specific TME conditions (pH, enzymes, redox status) offers promising avenues for enhanced specificity and reduced off-target effects [29] [27].
For researchers and drug development professionals, these findings highlight the necessity of incorporating robust TME models throughout the development pipeline, from initial design to preclinical evaluation. By fully acknowledging and addressing the complex biological realities of the tumor microenvironment, the field can advance more effective nanomedicine strategies with improved clinical translation potential.
Diagram 2: Integrated Research Pipeline for TME-Informed Nanoparticle Development. This workflow outlines a systematic approach to nanomedicine design that incorporates TME considerations at each stage, from initial barrier analysis through clinical translation, emphasizing the importance of physiologically relevant models.
Advanced Targeting Strategies and Nanoplatform Engineering
Conventional cancer treatments, including chemotherapy and radiotherapy, are significantly limited by systemic toxicity, limited tumor specificity, and the development of therapy resistance [34]. Targeted nanotherapy has emerged as a promising strategy to overcome these challenges by enhancing drug delivery precision to tumor sites. Ligand-functionalized nanoparticles represent a sophisticated class of drug delivery systems designed to recognize and bind specific receptors overexpressed on cancer cells, thereby improving therapeutic efficacy while reducing off-target effects [35] [36]. The strategic application of targeting ligands—including antibodies, peptides, aptamers, and transferrin—enables active targeting that surpasses the passive accumulation achieved through the Enhanced Permeability and Retention (EPR) effect alone [37]. This guide provides a comprehensive comparative analysis of these four prominent ligand classes, offering researchers in drug development a detailed examination of their performance characteristics, experimental methodologies, and applications within cancer therapeutics.
Performance Comparison of Targeting Ligands
The selection of an appropriate targeting ligand requires careful consideration of multiple performance parameters. The following table summarizes key characteristics of antibodies, aptamers, peptides, and transferrin to facilitate direct comparison.
Table 1: Comparative Performance of Ligands for Nanoparticle Functionalization
| Ligand Type | Targeting Mechanism | Affinity | Size | Immunogenicity | Production Complexity | Cost | Stability | Conjugation Chemistry |
|---|---|---|---|---|---|---|---|---|
| Antibodies | Specific antigen recognition [38] | High (pM-nM) [38] | Large (~150 kDa) | Moderate to High [39] | High [37] | High [39] | Moderate [39] | Site-specific (e.g., glycosite engineering) [37] |
| Aptamers | 3D structure-based binding [40] [39] | High (nM-pM) [41] | Small (~15 kDa) | Low [41] [39] | Moderate (SELEX) [40] | Moderate [39] | High (chemical modification possible) [40] [39] | Covalent (thiol, amine) [39] |
| Peptides | Receptor-ligand interaction [35] | Variable (nM-μM) | Small (~1-10 kDa) | Low to Moderate [35] | Low (chemical synthesis) | Low | Moderate | Covalent (amine, carboxyl) |
| Transferrin | Transferrin receptor binding [36] | Moderate (μM) | Medium (~80 kDa) | Low (endogenous) | Low (purification) | Low | High | Covalent (amine) |
Table 2: Experimental Performance Data Across Cancer Models
| Ligand Type | Nanoparticle Platform | Cancer Model | Target Receptor | Cellular Uptake Enhancement | Tumor Growth Inhibition | Reference |
|---|---|---|---|---|---|---|
| Anti-EGFR Antibody | PLGA-PEG | Pancreatic Cancer | EGFR | 3.2-fold increase vs. non-targeted NP [38] | 67% reduction vs. 42% for non-targeted NP [38] | [38] |
| RLA01 Aptamer | PLGA | Ovarian Cancer (Caov-3 cells) | Not Specified | 20-85% increase in cellular uptake [41] | Significant decrease in cell proliferation; induced apoptosis [41] | [41] |
| Transferrin | Gold NPs | Glioblastoma | Transferrin Receptor | 4.5-fold increase vs. non-targeted NP [36] | Improved survival in murine models [36] | [36] |
Ligand-Specific Experimental Protocols and Outcomes
Antibody-Functionalized Nanoparticles
Experimental Protocol: Antibody-Conjugated Polymer Nanoparticles for Brain Cancer
Antibody-conjugated nanoparticles require precise fabrication and conjugation methodologies. A representative protocol for creating antibody-functionalized polymer nanoparticles for brain cancer treatment involves several critical stages [42]:
Nanoparticle Synthesis: Polymeric nanoparticles (e.g., PLGA, PLA) are typically formulated using emulsion-solvent evaporation methods. Briefly, the polymer and hydrophobic drug are dissolved in an organic solvent (e.g., dichloromethane) and emulsified in an aqueous phase containing a stabilizer (e.g., polyvinyl alcohol) using probe sonication. The emulsion is then stirred overnight to evaporate the organic solvent, yielding solid nanoparticles [42].
Surface Functionalization: The nanoparticle surface is activated for antibody conjugation. A common approach introduces carboxylic acid groups (-COOH) on the nanoparticle surface, which are then activated with EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) and NHS (N-hydroxysuccinimide) to form amine-reactive esters.
Antibody Conjugation: The activated nanoparticles are incubated with the antibody solution (e.g., anti-EGFR, anti-HER2) under gentle agitation at 4°C for 12-24 hours. The unconjugated antibodies are removed by centrifugation and washing. The number of antibodies per nanoparticle can be quantified using techniques like the BCA assay or fluorescence labeling [37] [42].
In Vitro Validation: Cellular uptake is evaluated using flow cytometry and confocal microscopy in receptor-positive cancer cell lines. For instance, anti-EGFR antibody-conjugated nanoparticles showed significantly higher internalization in EGFR-overexpressing glioblastoma cells compared to isotype-controlled nanoparticles [42].
In Vivo Evaluation: Orthotopic tumor-bearing models (e.g., murine glioblastoma models) are used to assess targeting efficacy and therapeutic outcomes. Biodistribution studies often utilize fluorescence imaging or radiolabeling to demonstrate enhanced tumor accumulation of antibody-functionalized nanoparticles compared to their non-targeted counterparts [42].
Key Outcomes: Antibody-conjugation significantly enhances nanoparticle specificity. Preclinical studies demonstrate that anti-EGFR antibody-functionalized nanoparticles achieve up to 3.2-fold higher cellular uptake in EGFR-positive cancer cells compared to non-targeted controls and can reduce tumor growth by up to 67% in murine models [38] [42].
Aptamer-Functionalized Nanoparticles
Experimental Protocol: Aptamer-Coated PLGA Nanoparticles for Ovarian Cancer
The development of aptamer-labeled nanoparticles involves aptamer selection and conjugation, as exemplified by RLA01 aptamer-functionalized PLGA nanoparticles for ovarian cancer targeting [41]:
Aptamer Selection: The RLA01 aptamer was identified against epithelial ovarian cancer (EOC) Caov-3 cells using the Cell-SELEX (Systematic Evolution of Ligands by EXponential Enrichment) process. This iterative process involves incubation of a random single-stranded DNA library with target cells, removal of unbound sequences, elution of bound sequences, and amplification by PCR to enrich for high-affinity binders [41] [40].
Aptamer Modification: Selected aptamers are typically synthesized with chemical modifications (e.g., 2'-fluoropyrimidines) to enhance nuclease resistance and a terminal functional group (e.g., thiol, amine) for conjugation [40].
Nanoparticle Formulation and Conjugation: Paclitaxel-loaded PLGA nanoparticles are prepared using a single emulsion-solvent evaporation technique. For conjugation, aptamers are reduced (if thiol-modified) and incubated with maleimide-functionalized PLGA nanoparticles. The conjugation reaction proceeds for 12-24 hours at room temperature, followed by purification to remove unreacted aptamers [41].
Targeting Validation: Selective targeting is confirmed using co-culture assays with target (Caov-3) and non-target (HOSE 6-3) cells. RLA01-labeled nanoparticles exhibited 20-85% enhanced uptake in Caov-3 cells while simultaneously inhibiting uptake in non-target HOSE cells, demonstrating high selectivity [41].
Efficacy Assessment: The therapeutic efficacy of aptamer-targeted, drug-loaded nanoparticles is evaluated using cell proliferation assays (e.g., MTT) and apoptosis assays (e.g., Annexin V staining). RLA01-PLGA-Ptx significantly decreased cell proliferation and induced apoptosis in target cells [41].
Key Outcomes: Aptamer RLA01 facilitated selective internalization of PLGA nanoparticles into target EOC cells via receptor-mediated endocytosis, enhancing cellular uptake by 20-85% and significantly increasing paclitaxel-induced apoptosis. In vivo, aptamer labeling promoted nanoparticle retention at tumor sites [41].
Peptide-Functionalized Nanoparticles
Experimental Protocol: Peptide-Tagged Nanoparticles for Tumor Targeting
Peptide-functionalized nanoparticles leverage specific peptide-receptor interactions for targeted delivery. While search results provide limited specific protocol details for peptides, standard methodologies are well-established [35]:
Peptide Selection and Design: Targeting peptides (e.g., RGD for αvβ3 integrin) are selected based on known receptor overexpression in specific cancer types. Peptides can be identified through phage display libraries or designed from natural protein sequences.
Nanoparticle Preparation and Functionalization: Nanoparticles are synthesized from various materials (polymers, lipids, inorganic). Peptides are conjugated to the nanoparticle surface using carbodiimide chemistry, click chemistry, or maleimide-thiol reactions, often via PEG spacers to improve ligand accessibility.
Evaluation of Targeting Efficiency: Binding specificity and cellular uptake are assessed in receptor-positive versus receptor-negative cell lines. Internalization is often quantified using flow cytometry and visualized via confocal microscopy.
Key Outcomes: Peptide ligands effectively enhance nanoparticle accumulation in target tissues. For instance, RGD-functionalized nanoparticles demonstrate improved targeting to tumor vasculature and can penetrate deeper into tumor tissue compared to non-targeted systems, though their affinity is generally lower than that of antibodies or aptamers [35].
Transferrin-Functionalized Nanoparticles
Experimental Protocol: Transferrin-Conjugated Nanoparticles for Receptor-Mediated Uptake
Transferrin functionalization exploits the overexpression of transferrin receptors (TfR) on many cancer cells to facilitate nanoparticle internalization [36]:
Nanoparticle Synthesis: Metallic (e.g., gold, iron oxide) or polymeric nanoparticles are synthesized using standard chemical methods.
Transferrin Conjugation: Human transferrin is conjugated to activated nanoparticles (e.g., carboxylated or maleimide-functionalized surfaces) via amide or thioether bonds. The reaction mixture is purified via dialysis or centrifugation.
Cellular Uptake Studies: Targeting efficacy is validated using TfR-positive cancer cells. Competitive inhibition assays with free transferrin confirm receptor-mediated uptake.
In Vivo Biodistribution: Tumor targeting efficiency is evaluated in animal models, demonstrating significantly higher accumulation of transferrin-conjugated nanoparticles in tumors compared to non-targeted controls.
Key Outcomes: Transferrin-functionalized nanoparticles achieve up to 4.5-fold higher uptake in TfR-overexpressing cancer cells compared to non-targeted nanoparticles and can improve survival in murine glioma models [36].
Visualizing Experimental Workflows and Targeting Mechanisms
Aptamer-Nanoparticle Conjugation and Targeting Workflow
Diagram 1: Aptamer Selection and NP Conjugation. This workflow outlines the process from aptamer identification through Cell-SELEX to conjugation with nanoparticles and final validation.
Ligand-Receptor Signaling and Internalization Pathways
Diagram 2: NP Targeting and Internalization Pathway. This diagram illustrates the sequential process from ligand-receptor binding to internalization and intracellular drug release.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Developing Ligand-Functionalized Nanoparticles
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Nanoparticle Polymers | PLGA, PLA, PEG, Chitosan [41] [35] | Nanoparticle matrix formation | Biodegradable backbone for drug encapsulation and controlled release [35] |
| Targeting Ligands | Anti-EGFR Antibody [38], RLA01 Aptamer [41], RGD Peptide, Transferrin [36] | Active tumor targeting | Bind overexpressed receptors on cancer cells for specific cellular uptake [37] [36] |
| Conjugation Reagents | EDC, NHS, Maleimide, Click Chemistry reagents (e.g., DBCO, Azides) [37] | Covalent ligand attachment to NPs | Facilitate stable bond formation between nanoparticle surface and targeting ligand [37] |
| Characterization Tools | DLS, Zeta Potential Analyzer, TEM/SEM, HPLC | NP physicochemical characterization | Determine size, surface charge, morphology, and drug loading efficiency |
| Cell Culture Models | Caov-3 (Ovarian) [41], Glioblastoma lines (U87, U251) [40] [42], MCF-7 (Breast) | In vitro targeting and efficacy studies | Provide receptor-positive cellular models for validating targeting specificity and therapeutic response |
| Animal Models | Orthotopic xenografts, Genetically engineered models (GEMMs), Patient-derived xenografts (PDX) [41] [42] | In vivo biodistribution and efficacy | Recapitulate human tumor microenvironment and biology for preclinical testing |
The strategic functionalization of nanoparticles with targeting ligands represents a cornerstone of precision oncology, dramatically improving the specificity and efficacy of cancer therapeutics. Each ligand class offers distinct advantages: antibodies provide high specificity and affinity, aptamers combine high stability and low immunogenicity, peptides offer simplicity and ease of production, and transferrin leverages a naturally occurring uptake pathway. The selection of an optimal ligand depends on the specific therapeutic context, including the target receptor, tumor type, and physiological barriers. As conjugation technologies advance and our understanding of tumor biology deepens, ligand-functionalized nanoparticles are poised to deliver increasingly sophisticated and effective cancer treatments, ultimately translating into improved patient outcomes. Future directions will likely focus on multi-ligand targeting systems and stimuli-responsive designs that further enhance targeting precision and therapeutic efficacy.
Targeted cancer therapies represent a cornerstone of modern oncology, aiming to maximize therapeutic efficacy while minimizing off-target effects. Receptor-mediated targeting leverages the overexpression of specific receptors on cancer cells to direct therapeutic agents precisely to tumor tissues. This guide provides a comparative analysis of four principal receptor systems—Epidermal Growth Factor Receptor (EGFR), Integrins, Folate Receptors (FR), and Transferrin Receptors (TfR)—evaluating their targeting efficiency, experimental applications, and suitability for nanoparticle-based drug delivery in cancer therapy research.
The utility of a receptor for targeted therapy is largely determined by its expression pattern across normal and cancerous tissues.
- EGFR is a receptor tyrosine kinase significantly overexpressed in cancers such as colorectal, lung, and head and neck cancers. Its signaling governs critical cellular processes including proliferation, survival, and metastasis [43] [44].
- Integrins, a family of heterodimeric transmembrane receptors, are upregulated on both tumor cells and the associated vasculature. Specific members like αvβ3, αvβ5, and α5β1 recognize the Arg-Gly-Asp (RGD) peptide motif, making them prime targets for peptide-directed delivery [45] [46].
- Folate Receptors (FRα/FRβ) are overexpressed in a wide range of cancers, including ovarian, lung, and breast cancers, while their expression in most normal tissues is limited. FRα is also a key marker on activated macrophages in inflammatory diseases like rheumatoid arthritis, broadening its therapeutic relevance [47] [48] [49].
- Transferrin Receptors (TfR1) are universally upregulated on rapidly dividing cells, including cancer cells, due to their increased demand for iron. This makes TfR1 a versatile target across many cancer types [43] [50].
The diagram below illustrates the foundational mechanism of receptor-mediated endocytosis shared by these targeting strategies.
Comparative Targeting Efficiency Analysis
The following tables synthesize experimental data from recent studies, providing a direct comparison of the targeting performance and therapeutic outcomes associated with each receptor system.
Table 1: Quantitative Comparison of Targeting Efficiencies
| Receptor | Targeting Ligand | Cancer Model | Key Performance Metric | Result | Reference |
|---|---|---|---|---|---|
| EGFR | Cetuximab (Antibody) | Colorectal (SW480) | Cellular Uptake (Targeted vs. Non-targeted NP) | Significantly Enhanced | [44] |
| Integrin αvβ3 | RGD Peptide | Various (Melanoma, Glioma) | Tumor Accumulation (RGD-functionalized carriers) | Improved Efficiency | [45] [46] |
| Folate Receptor (FRα) | FolTAC-dual (Bispecific) | HER2+ Breast Cancer | Protein Degradation (EGFR/HER2) | ~85% Efficiency | [47] |
| Folate Receptor (FRα) | Pemetrexed (Small Molecule) | Colorectal (CT26) | Tumor Growth Inhibition (Targeted vs. Non-targeted NP) | Significantly Greater | [51] |
| Folate Receptor (FRα) | FRTAC (Bispecific) | Syngeneic Mouse Models | Tumor Growth Suppression (vs. Blocking Antibody) | More Significant | [52] |
| Transferrin Receptor (TfR1) | TransTAC (Bispecific) | EGFR-mutant Lung Cancer | Membrane Protein Degradation | Efficient & Broad | [50] |
Table 2: Analysis of Advantages and Clinical Challenges
| Receptor | Key Advantages | Primary Challenges & Considerations |
|---|---|---|
| EGFR | Well-established target; FDA-approved ligands (e.g., Cetuximab); strong internalization. | Heterogeneous expression; potential for signaling-mediated resistance. |
| Integrins | Accessible via simple RGD peptides; targets both tumor cells and angiogenic vasculature. | Complexity from multiple heterodimers; achieving subtype selectivity is difficult. |
| Folate Receptors | High affinity for folate; high selectivity for cancerous/activated cells; non-immunogenic. | Expression can vary by cancer type; potential competition from circulating folate. |
| Transferrin Receptors | Very high expression on most cancer types; rapid internalization cycle. | High background in normal proliferative tissues; potential competition from endogenous transferrin. |
Experimental Protocols for Key Studies
To facilitate experimental replication and development, this section details the methodologies behind pivotal studies for each receptor.
EGFR-Targeted Chemo-Photothermal Therapy
This protocol is adapted from the development of Cetuximab-targeted, Irinotecan and ICG-loaded nanoparticles (Cet-Iri-NPs) for colorectal cancer [44].
- Nanoparticle Formulation: Lipid-polymer hybrid nanoparticles (Cet-Iri-NPs) were prepared using a single-step ultrasonic emulsion method. The copolymer PPG-PEG, lipids (lecithin, DSPE-PEG-Mal), CPT-11 (Irinotecan), and the photothermal agent Indocyanine Green (ICG) were combined in an aqueous suspension under sonication.
- Ligand Conjugation: Cetuximab was first thiolated using Traut's reagent. The thiolated antibody was then conjugated to maleimide-functionalized nanoparticles (Iri-NPs) via a thiol-maleimide click reaction at a molar ratio of 1:100 in PBS (pH 8.5-9.0) at 4°C for 24 hours.
- In Vitro Cytotoxicity Assay: SW480 human colorectal cancer cells were treated with free CPT-11, non-targeted Iri-NPs, or targeted Cet-Iri-NPs, with and without NIR laser irradiation. Cell viability was assessed using a standard Cell Counting Kit-8 (CCK-8) after incubation.
- In Vivo Efficacy Study: SW480 tumor xenograft models in nude mice were established. Mice were intravenously administered with different formulations, and tumor dimensions were measured regularly with calipers to calculate tumor volume. Mice in the photothermal therapy groups were exposed to NIR laser irradiation at the tumor site post-injection.
Integrin Targeting with RGD Peptides
This methodology outlines the use of RGD peptides for targeted drug delivery, as reviewed in recent literature [45] [46].
- Ligand Functionalization: RGD peptides or their analogs (e.g., cyclic RGDfK) are synthetically conjugated to the surface of nanocarriers (e.g., polymeric NPs, liposomes) via covalent attachment to functional groups on PEGylated lipids or polymers.
- Cellular Uptake and Binding Assay: Integrin-overexpressing cancer cells (e.g., U87MG for αvβ3) are incubated with RGD-functionalized carriers, often loaded with a fluorescent dye (e.g., DiR, Coumarin-6). Uptake is quantified using flow cytometry and visualized with confocal microscopy. Specificity is confirmed via competitive inhibition with free RGD peptide.
- In Vivo Imaging and Biodistribution: Tumor-bearing mice are injected intravenously with RGD-conjugated imaging agents (e.g., fluorescent probes or radiotracers). Whole-body imaging is performed over time using systems like IVIS for fluorescence or SPECT/CT for radioactivity to evaluate tumor accumulation and biodistribution.
Folate Receptor-Mediated Targeted Degradation (FolTAC)
This protocol describes the engineering and evaluation of bispecific degraders for dual membrane protein degradation [47].
- Degrader Construction: FolTAC-dual molecules are engineered on an antibody scaffold. The "string" configuration format is utilized, where optimized receptor-targeting binders (e.g., affibodies or scFvs for EGFR and HER2) are connected via modular linkers to a Folate Receptor α (FRα)-targeting unit (e.g., Farletuzumab scFv).
- Binding Affinity Measurement: Surface Plasmon Resonance (SPR) or similar biophysical techniques are used to measure the binding affinity (KD) of the FolTAC constructs to target receptors (e.g., EGFR). This is compared to conventional designs like knob-into-hole to validate affinity improvements.
- Degradation Efficiency Assay: Target cancer cells (e.g., HER2-positive breast cancer cells) are treated with FolTAC-dual degraders. Protein levels of the targets (e.g., EGFR and HER2) are quantified over time (e.g., 24-48 hours) via Western blotting or flow cytometry to determine degradation efficiency.
- In Vivo Resistance Models: Proof-of-concept studies are conducted in mouse models of therapy-resistant cancer, such as Trastuzumab/Lapatinib-resistant HER2-positive breast cancer. Tumor growth inhibition is monitored in mice treated with FolTAC-dual versus control therapies.
Transferrin Receptor-Mediated Degradation (TransTAC)
This protocol summarizes the application of Transferrin Receptor Targeting Chimeras for membrane protein degradation [50].
- TransTAC Engineering: Heterobispecific antibodies (TransTACs) are designed with one binding arm for TfR1 and another for the specific Protein of Interest (POI), which can be a single-pass or multi-pass membrane protein.
- In Vitro Degradation Profiling: A panel of cell lines expressing different target proteins (e.g., EGFR, PD-L1, CD20) is treated with TransTACs. The extent of target protein degradation is assessed using flow cytometry or Western blot after a defined incubation period.
- In Vivo Efficacy in Resistant Models: The technology is tested in vivo using mouse xenograft models of drug-resistant cancers, such as EGFR-driven lung cancer harboring the exon 19 deletion/T790M/C797S mutations. Tumor volume is tracked in animals treated with TransTACs versus standard-of-care agents.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Receptor-Targeting Research
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Cetuximab | Monoclonal antibody targeting EGFR; used for conjugation to NPs. | Active targeting of EGFR-overexpressing cancers like CRC [44]. |
| RGD Peptide (cyclic) | High-affinity integrin-binding ligand; enables functionalization of carriers. | Targeting αvβ3/αvβ5 integrins on tumor cells and vasculature [45]. |
| Folic Acid / Pemetrexed | High-affinity ligands for Folate Receptor (FRα); used as targeting moieties. | Synthesizing FR-targeted polymers for NP drug delivery [51]. |
| Folate Conjugates (e.g., DBCO-PEG3-NHS, Azido-PEG-Folate) | Commercial kits for facile conjugation of folate to antibodies or proteins. | Generating bispecific Folate Receptor TArgeting Chimeras (FRTACs) [52]. |
| PLGA-PEG Copolymer | Biodegradable polymer for NP formulation; PEG provides stealth properties. | Core material for constructing targeted nanoparticles [51]. |
| DSPE-PEG-Mal | Amphiphilic lipid; anchors in NP lipid layer while providing maleimide group for ligand conjugation. | Conjugating thiolated antibodies (e.g., Cetuximab) to nanoparticles [44]. |
| Indocyanine Green (ICG) | FDA-approved photothermal agent; generates heat under NIR irradiation. | Enabling combination chemo-photothermal therapy in targeted NPs [44]. |
| Tin(II) 2-ethylhexanoate (Sn(Oct)₂) | Catalyst for ring-opening polymerization (ROP). | Synthesizing custom PLGA-PEG-pemetrexed polymers [51]. |
The choice of receptor system for targeted cancer therapy is contingent on the specific research and clinical context. EGFR provides a well-validated pathway with clinically available targeting agents. Integrin targeting via RGD peptides offers a versatile platform for targeting both tumor and stromal cells. Emerging bispecific degradation technologies, TransTACs (TfR) and FolTACs/FRTACs (FR), demonstrate powerful and broad capabilities for directly eliminating pathogenic membrane proteins. Ultimately, the selection depends on the tumor type's receptor expression profile, the desired mechanism of action (inhibition vs. degradation), and the physicochemical properties of the therapeutic cargo. A deep understanding of these systems' efficiencies, advantages, and limitations, as detailed in this guide, is fundamental to advancing more effective and precise cancer nanomedicines.
The efficacy of conventional cancer therapeutics is often limited by their inability to distinguish effectively between healthy and malignant cells, leading to systemic toxicity and narrow therapeutic windows. Stimuli-responsive nanosystems, often termed "smart" drug delivery systems, represent a paradigm shift in oncology by enabling precise spatiotemporal control over therapeutic release. These nanocarriers are engineered to remain stable in circulation but undergo triggered drug release upon encountering specific physiological or externally applied stimuli at the tumor site. This capability is critically framed within the broader challenge of nanoparticle targeting efficiency in cancer therapy, which relies on a multi-stage process involving circulation, accumulation, penetration, internalization, and intracellular trafficking. By responding to distinctive features of the tumor microenvironment (TME) or external triggers, these systems directly address inefficiencies at nearly every stage of targeting, offering a powerful strategy to enhance therapeutic index. This guide provides a comparative analysis of four major stimulus categories—pH, redox potential, temperature, and enzymes—objectively evaluating their mechanisms, performance, and experimental validation to inform research and development.
Comparative Mechanisms of Stimuli-Responsive Nanosystems
The tumor microenvironment possesses unique biochemical and physical characteristics that differ from healthy tissues. Stimuli-responsive nanosystems are designed to exploit these differences. Table 1 provides a comprehensive comparison of the four primary stimuli-responsive nanosystems, detailing their triggering mechanisms, representative materials, and key performance metrics.
Table 1: Comparative Analysis of Stimuli-Responsive Nanosystems in Cancer Therapy
| Stimulus Type | Trigger Mechanism in TME | Responsive Chemical Groups/Materials | Drug Release Mechanism | Key Performance Metrics (from experimental data) |
|---|---|---|---|---|
| pH-Sensitive | Exploits acidic tumor microenvironment (pH ~6.5-6.8) and endo/lysosomal compartments (pH ~4.5-5.0) [53]. | Polyhistidine, poly(acrylic acid), acetal, ketal, hydrazone bonds [53] [54]. | Protonation-induced charge shift, cleavage of acid-labile bonds, or disruption of hydrophobic interactions leading to nanocarrier disassembly [53]. | > 80% drug release at pH 5.0 vs. < 20% at pH 7.4 within 24-48 hours; demonstrated enhanced cytotoxicity in cancer cells [53]. |
| Redox-Sensitive | Leverages high intracellular glutathione (GSH) concentration (2-10 mM in cytosol vs. 2-20 μM in plasma) [55]. | Disulfide (S-S), diselenide (Se-Se) bonds; thioether, selenide-containing polymers [56] [55] [57]. | Thiol-disulfide exchange or redox-induced solubility change (hydrophobic to hydrophilic), causing nanoparticle destabilization and cargo release [55] [57]. | Near-complete drug release (>90%) within hours in 10 mM GSH; GSH depletion can synergistically induce ferroptosis/cuproptosis [55]. |
| Temperature-Sensitive | Utilizes mildly elevated temperature in tumors (~40°C) or externally applied heat [58]. | Poly(N-isopropylacrylamide) (PNIPAAm), pluronics (PEO-PPO), elastin-like polypeptides (ELPs) with LCST behavior [58]. | Phase transition (swelling/collapse) of polymers at Lower Critical Solution Temperature (LCST), leading to burst drug release [58]. | Significant drug release increase (2- to 5-fold) upon heating from 37°C to 40-42°C; enables hyperthermia-mediated combination therapy [58]. |
| Enzyme-Sensitive | Responds to overexpressed enzymes in TME (e.g., MMPs, cathepsins, phosphatases) [54]. | Enzyme-specific cleavable peptide linkers (e.g., GFLG for cathepsin B, PLGLAG for MMP-2) [55] [54]. | Enzymatic hydrolysis of specific linkers, de-PEGylation, or cleavage of prodrugs to activate therapeutics [54]. | Site-specific drug activation and release; demonstrated protease-activatable cell penetration [54]. |
The following diagram illustrates the core mechanisms and sequential drug release process shared by these stimuli-responsive nanosystems, from initial injection to intracellular drug release.
Diagram 1: Generalized mechanism of stimuli-responsive drug release, showing the common pathway from administration to therapeutic effect, triggered by specific TME signals.
Experimental Protocols for Evaluating Stimuli-Responsive Release
To objectively compare the performance of different stimuli-responsive nanosystems, standardized experimental protocols are essential. Below are detailed methodologies for evaluating release profiles and targeting efficiency, as cited in recent literature.
In Vitro Drug Release Kinetics
Objective: To quantify the rate and extent of drug release from nanocarriers under simulated physiological (normal) and pathological (stimulus) conditions.
Protocol for pH-Responsive Release [53]:
- Preparation: Dialyze a known quantity of drug-loaded nanocarriers against phosphate-buffered saline (PBS) at pH 7.4 (mimicking blood) and pH 5.0 or 6.5 (mimicking tumor microenvironment or endosomes) at 37°C under constant agitation.
- Sampling: At predetermined time intervals, withdraw a sample from the release medium and replace it with an equal volume of fresh buffer to maintain sink conditions.
- Analysis: Quantify the drug concentration in the samples using a validated analytical method, such as High-Performance Liquid Chromatography (HPLC) or fluorescence spectroscopy, depending on the drug's properties.
- Data Processing: Calculate the cumulative drug release percentage over time. Plot the release profile to compare kinetics at different pH levels.
Protocol for Redox-Responsive Release [55]:
- Follow the general dialysis procedure above.
- Stimulus Introduction: Use PBS buffers containing 10 μM GSH (mimicking extracellular levels) and 10 mM GSH (mimicking intracellular levels) as the release media.
- Monitor and compare the drug release profiles to confirm the redox-triggered disassembly of disulfide- or diselenide-based nanocarriers.
Key Performance Metric: The Stimulus Response Ratio (SRR), calculated as (Cumulative Release at Stimulus Condition) / (Cumulative Release at Normal Condition) at a specific endpoint (e.g., 24 hours). A higher SRR indicates greater specificity.
In Vitro Cellular Uptake and Targeting Efficiency
Objective: To validate that active targeting moieties enhance nanoparticle internalization by target cancer cells and to assess the role of stimuli in intracellular drug release.
Protocol for RGD-Functionalized Gold Nanoparticles (GNPs) [25]:
- Cell Culture: Seed cancer cells (e.g., KPCY murine pancreatic cancer cells) known to overexpress the target receptor (ανβ3 integrin) in monolayer or 3D spheroid cultures.
- Nanoparticle Exposure: Incubate cells with targeted (e.g., GNP-PEG-cRGD) and non-targeted (e.g., GNP-PEG) nanoparticles at a consistent concentration (e.g., 7.5 µg/mL) for varying durations (1h, 8h, 24h).
- Quantification (ICP-MS): After incubation, wash the cells thoroughly to remove non-internalized nanoparticles. Lyse the cells and analyze the lysate using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) to precisely quantify the gold content, which corresponds to the number of internalized nanoparticles.
- Qualitative Confirmation: Use confocal laser scanning microscopy (CLSM) to visually confirm the intracellular localization of fluorescently labeled nanoparticles.
Key Performance Metric: Targeting Enhancement Factor, calculated as (Cellular uptake of targeted NPs) / (Cellular uptake of non-targeted NPs). In the cited study, cRGD-functionalized GNPs showed a 150-fold increase in uptake at 1 hour compared to non-targeted GNPs [25].
Signaling Pathways and Therapeutic Synergy
Redox-manipulating nanosystems offer a unique therapeutic advantage by not only releasing drugs but also actively disrupting the redox homeostasis of cancer cells. This dual action can trigger specific cell death pathways, as illustrated below.
Diagram 2: Signaling pathways of GSH depletion-induced ferroptosis and cuproptosis, showcasing the therapeutic synergy of redox-responsive nanosystems.
The pathway shows how glutathione (GSH) depletion by nanocarriers [55] inhibits the glutathione peroxidase 4 (GPX4) enzyme, leading to toxic lipid peroxide accumulation and ferroptosis. Concurrently, GSH depletion promotes dihydrolipoamide S-acetyltransferase (DLAT) oligomerization, sensitizing cells to cuproptosis [55]. This represents a sophisticated therapeutic strategy beyond simple drug delivery.
The Scientist's Toolkit: Key Research Reagents and Materials
The development and evaluation of stimuli-responsive nanosystems rely on a specific set of chemical and biological reagents. Table 2 catalogs essential materials used in the featured experiments and the broader field.
Table 2: Essential Research Reagents for Stimuli-Responsive Nanosystems
| Reagent/Material | Function in Research | Specific Example & Context |
|---|---|---|
| Thiol-Terminated PEG (e.g., mPEG-SH) | Confers "stealth" properties, prolongs circulation, provides anchor for conjugation. | Used at 1 PEG/nm² on gold nanoparticles (GNPs) to minimize protein adsorption [25]. |
| RGD Peptides (linear & cyclic) | Active targeting ligand for integrin ανβ3, promotes cellular internalization. | cRGD and lRGD grafted at 1:2 ratio (RGD:PEG) on GNPs to assess targeting efficacy [25]. |
| Poly(N-isopropylacrylamide) (PNIPAAm) | Thermoresponsive polymer with LCST ~32°C; backbone of temperature-sensitive systems. | Used in hydrogels and core-shell NPs for heat-triggered drug release [58]. |
| Disulfide Crosslinkers (e.g., cystamine) | Forms redox-responsive bonds in polymer backbones or between drug and carrier. | Critical for constructing nanocarriers that degrade in high intracellular GSH [55]. |
| GSH (Reduced Glutathione) | Key reagent for creating reducing environments in vitro to mimic intracellular conditions. | Used at 10 mM concentration in release media to trigger redox-sensitive drug release [55]. |
| Citrate-Capped Gold Nanoparticles (GNPs) | Versatile, inert inorganic nanocarrier core; easy surface functionalization via thiol chemistry. | ~12 nm spherical GNPs used as a model platform to study RGD targeting efficiency [25]. |
The objective comparison of pH, redox, temperature, and enzyme-responsive nanosystems reveals a trade-off between the specificity of the trigger and its clinical applicability. While pH and redox-responsive systems leverage inherent TME features and show high specificity in vitro, their efficacy in vivo can be heterogeneous due to patient-specific variations in the EPR effect and TME composition. Temperature-responsive systems offer excellent external control but require specialized equipment for heat application. Enzyme-responsive systems provide high specificity but depend on the consistent and selective overexpression of the target enzyme.
The future of stimuli-responsive nanosystems lies in the development of multi-responsive platforms that integrate two or more triggers (e.g., pH/redox, thermo/enzyme) to enhance specificity and control. Furthermore, the integration of biomimetic strategies (e.g., cell membrane coating) and advanced technologies like AI-driven design and tumor-on-chip models for testing will be crucial for overcoming translational hurdles. The ultimate goal remains the rational design of nanocarriers that can successfully navigate the biological barriers to achieve efficient multi-stage targeting, from systemic circulation to precise subcellular drug release, thereby fully realizing the potential of nanomedicine in oncology.
The efficacy of conventional cancer therapeutics is often limited by their inability to distinguish effectively between healthy and cancerous cells, leading to systemic toxicity and dose-limiting side effects. Within the broader thesis of improving nanoparticle targeting efficiency in cancer therapy research, externally activated platforms represent a paradigm shift, offering unprecedented spatial and temporal control over treatment delivery. These sophisticated systems respond to physical stimuli—magnetic fields, light, or ultrasound—applied from outside the body, enabling precise triggering of therapeutic effects specifically at the tumor site [34] [59].
Magnetic nanoparticles (MNPs) leverage external magnetic fields for guidance and activation, while light-responsive systems exploit specific wavelengths to initiate cytotoxic processes. Ultrasound-responsive nanocarriers utilize mechanical and thermal effects from sound waves for enhanced tissue penetration and drug release [59] [60]. The fundamental advantage shared by these platforms is their ability to remain relatively inert in circulation until activated by their respective external trigger at the target site, thereby maximizing therapeutic impact on tumors while minimizing off-target effects on healthy tissues—a critical advancement in targeted cancer therapy research [61] [62].
Comparative Performance Analysis of Externally Activated Platforms
The table below provides a systematic comparison of the three major classes of externally activated nanoparticle platforms, highlighting their key characteristics, performance metrics, and research applications.
Table 1: Comparative Analysis of Externally Activated Nanoparticle Platforms
| Platform Feature | Magnetic Nanoparticles | Light-Responsive Nanoparticles | Ultrasound-Responsive Nanocarriers |
|---|---|---|---|
| Activation Mechanism | Alternating magnetic fields (hyperthermia), static fields (guidance) | Near-infrared (NIR) light, UV/visible light (limited use) | High/low-intensity focused ultrasound (HIFU/LIFU) |
| Primary Applications | Magnetic hyperthermia, targeted drug delivery, MRI contrast | Photothermal therapy (PTT), photodynamic therapy (PDT), triggered drug release | Enhanced drug penetration, triggered release, hyperthermia |
| Tissue Penetration Depth | Unlimited for magnetic fields | Several centimeters (NIR light) | Unlimited (deep tissue capability) |
| Key Performance Metrics | • Heating efficiency (SAR)• Magnetic guidance accuracy• Imaging contrast enhancement | • Photothermal conversion efficiency• Reactive oxygen species (ROS) generation• Light-triggered release kinetics | • Drug release efficiency• Cavitation effects• Thermal enhancement ratio |
| Spatial Resolution | Moderate (millimeter-scale) | High (micrometer-scale with focusing) | Moderate to high (millimeter-scale with focusing) |
| Representative Nanoparticle Types | Iron oxide NPs, magnetic ionic liquids [63], ferromagnetic alloys | Gold NPs, carbon nanohorns [63], conjugated polymers [64] | Micro/nanobubbles, liposomes, polymeric micelles [59] [60] |
| Therapeutic Payload | Drugs, genes, hyperthermia | Drugs, photosensitizers, immunotherapeutics [64] | Chemotherapeutics, genes, sonosensitizers |
| Clinical Translation Status | Multiple clinical trials, some approved agents | Several in clinical trials, primarily for superficial tumors | Approved formulations (e.g., thermosensitive liposomes) [60] |
Table 2: Quantitative Experimental Data from Recent Studies
| Study Reference | Nanoparticle Type | Activation Method | Therapeutic Outcome | Tumor Model |
|---|---|---|---|---|
| Miyako et al. [63] | Magnetic carbon nanohorns | NIR laser (808 nm) + magnet | Complete tumor elimination after 6 treatments; no recurrence in 20 days | Mouse colon carcinoma (Colon26) |
| Tsourkas et al. [65] | Magnetic nanoclusters with Ce6 | 8-magnet array + red laser | 3.7× more particles, 3.5× deeper penetration; significant tumor growth delay | Mouse triple-negative breast cancer |
| Achilefu et al. [66] | Titanocene-loaded targeted NPs | PET imaging agent (FDG) as light source | 50% survival at 90 days vs. 62 days in controls; significant tumor reduction | Mouse multiple myeloma and metastatic breast cancer |
| Lyon et al. [60] | Thermosensitive liposomes | Focused ultrasound (FUS) | Successful human proof of concept for liver and pancreatic malignancies | Human liver and pancreatic cancers |
Magnetic Nanoparticles: Mechanisms and Experimental Approaches
Fundamental Mechanisms
Magnetic nanoparticles for cancer therapy primarily operate through two distinct mechanisms: magnetic hyperthermia and magnetic guidance. In magnetic hyperthermia applications, superparamagnetic iron oxide nanoparticles (SPIONs) or similar materials generate localized heat when subjected to an alternating magnetic field (AMF). This thermal energy elevates tumor temperature to the critical 42-45°C range, inducing apoptosis in cancer cells while sparing healthy tissues due to their higher thermal tolerance [61]. The second mechanism involves using static magnetic fields to guide drug-loaded magnetic nanoparticles to specific target sites, enhancing accumulation at tumor locations through physical manipulation [65].
The heating efficiency of magnetic nanoparticles is quantified by specific absorption rate (SAR), which depends on multiple factors including nanoparticle size, composition, coating, and magnetic field parameters (frequency and amplitude). Core-shell structures with iron oxide cores and biocompatible polymer shells (e.g., polyethylene glycol, chitosan) represent common configurations that provide both functionality and improved bioavailability [61].
Experimental Protocols and Key Findings
A recent groundbreaking study by Professor Miyako's team demonstrated a novel approach using magnetic ionic liquid-modified carbon nanohorns [63]. The experimental workflow involved synthesizing spherical graphene-based carbon nanohorns (CNHs) approximately 120 nanometers in size, then modifying their surface with magnetic ionic liquid 1-butyl-3-methylimidazolium tetrachloroferrate ([Bmim][FeCl4]) to impart magnetic properties. To address hydrophobicity challenges, researchers added a polyethylene glycol (PEG) coating to improve water dispersibility and incorporated indocyanine green fluorescent dye for tracking.
In laboratory testing, these nanoparticles were added to mouse-derived colon carcinoma (Colon26) cells and exposed to an 808 nm NIR laser at 0.7 W for 5 minutes. The nanoparticles demonstrated an exceptional photothermal conversion efficiency of 63%, sufficient to kill cancer cells. For in vivo validation, mice with Colon26 tumors received injections of the nanoparticles, which were guided to the tumor site using an external magnet. Subsequent laser activation heated tumors to 56°C, completely eliminating them after six treatments with no recurrence over 20 days. Crucially, control experiments without magnetic guidance showed tumor regrowth after laser treatment cessation, highlighting the essential role of magnetic targeting in achieving sufficient nanoparticle accumulation for complete eradication [63].
A complementary study from the University of Pennsylvania addressed the challenge of poor nanoparticle penetration into dense tumor tissues [65]. Researchers developed an eight-magnet Halbach array system capable of generating stronger, multidirectional magnetic fields compared to conventional two-magnet setups. They coated clusters of magnetic nanoparticles with chlorin e6 (Ce6), a compound that generates toxic free radicals when activated by light.
In mouse models of triple-negative breast cancer, this system achieved 3.7 times greater nanoparticle accumulation and 3.5 times deeper penetration into tumors compared to previous methods. The magnetic guidance followed by laser activation of Ce6 significantly slowed tumor growth, demonstrating the potential of advanced magnetic systems to overcome penetration barriers in solid tumors [65].
Diagram 1: Magnetic Nanoparticle Experimental Workflow
Light-Responsive Nanoplatforms: Mechanisms and Experimental Approaches
Fundamental Mechanisms
Light-responsive nanoparticles utilize specific wavelengths of light to trigger therapeutic effects through various mechanisms, with photothermal therapy (PTT) and photodynamic therapy (PDT) representing the most prominent approaches. In PTT, nanoparticles such as gold nanostructures, carbon nanohorns, or conjugated polymers absorb light energy (typically near-infrared for its superior tissue penetration) and convert it to heat, inducing localized thermal damage to cancer cells [63] [64]. PDT operates through a different mechanism where photosensitizers (e.g., chlorin e6, porphyrins) generate cytotoxic reactive oxygen species (ROS) upon light activation, leading to oxidative damage and cell death [65] [66].
More advanced light-responsive systems incorporate sophisticated drug release mechanisms, including photocleavable linkers that break under illumination or nanovalves that open in response to light-induced structural changes. The convergence of phototherapy with immunotherapy represents a particularly promising frontier, as photothermal damage can stimulate immune responses against tumors by releasing tumor antigens and altering the immunosuppressive tumor microenvironment [64].
Experimental Protocols and Key Findings
A pioneering approach from Washington University School of Medicine addressed the challenge of treating metastatic cancer using a novel light-activated strategy [66]. Researchers packaged the chemotherapy drug titanocene into nanoparticles targeted to proteins on cancer cell surfaces. When these nanoparticles contacted cancer cells, their membranes fused, releasing titanocene into the cells. The innovative aspect involved using a common cancer imaging agent, fluorodeoxyglucose (FDG), which is preferentially taken up by energy-hungry cancer cells, causing tumors to glow during PET scans. This glow simultaneously activated the titanocene, releasing free radicals that killed the cancer cells.
In mouse models of multiple myeloma, this approach resulted in 50% of treated mice surviving at least 90 days compared to 62 days in control groups. The treatment also showed efficacy against aggressive metastatic breast cancer, though with less pronounced effects due to the extreme aggressiveness of the cancer line used. Notably, researchers discovered that resistant multiple myeloma cells lacked the surface proteins targeted by the nanoparticles, providing insights for overcoming treatment resistance through alternative targeting strategies [66].
Conjugated polymer nanoparticles (CPNs) represent another innovative light-responsive platform [64]. One study developed CPNs approximately 52nm in diameter that were functionalized with thiolated cyclo (Arg-Gly-Asp-D-Phe-Lys (mpa)) peptide (c-RGD) to improve targeting. Under 808nm laser illumination, these CPNs exhibited high photothermal conversion efficiency, effectively killing cancer cells while simultaneously activating a proinflammatory immune response. This dual functionality highlights the potential of light-responsive systems to combine direct tumor destruction with immune stimulation for enhanced therapeutic outcomes.
Diagram 2: Light Activation Mechanisms and Outcomes
Ultrasound-Responsive Nanoplatforms: Mechanisms and Experimental Approaches
Fundamental Mechanisms
Ultrasound-responsive nanocarriers leverage mechanical and thermal effects generated by ultrasound waves to achieve targeted drug release and enhanced tissue penetration. The primary mechanisms include acoustic cavitation (formation and oscillation of microbubbles), acoustic radiation force, and thermal effects from energy absorption [59] [60]. These physical phenomena can temporarily disrupt cellular membranes and vascular structures, increasing permeability and facilitating nanoparticle extravasation into tumor tissues.
Thermosensitive liposomes represent a particularly advanced category of ultrasound-responsive systems, designed to release their drug payload upon mild heating induced by ultrasound energy absorption [60]. Other platforms include micro/nanobubbles that oscillate or collapse under ultrasound exposure, generating mechanical forces that enhance drug penetration through biological barriers. The unique advantage of ultrasound lies in its exceptional tissue penetration capability, enabling non-invasive treatment of deeply seated tumors that are inaccessible to light-based therapies.
Experimental Protocols and Key Findings
Therapeutic ultrasound typically employs one of three approaches for drug delivery: low-intensity pulsed ultrasound (LIPUS) for large treatment areas, and focused ultrasound (LIFU and HIFU) for small, precise regions [60]. Parameters including transducer power, ultrasound pressure, exposure time, and duty cycle must be carefully optimized to maintain tissue temperature below the threshold for thermal damage (typically <44°C for hyperthermia applications).
In human clinical trials, TUS-activated nanosized drug delivery systems using thermosensitive liposomes have successfully treated liver and pancreatic malignancies [60]. These implementations demonstrate the clinical viability of ultrasound-responsive platforms, particularly for tumors that are challenging to treat with conventional approaches. The combination of ultrasound with magnetic resonance guidance (MRgFUS) further enhances treatment precision by enabling real-time monitoring of temperature and drug release.
The integration of ultrasound with nanotechnology addresses several critical challenges in cancer drug delivery, including poor uptake and accumulation of nanoparticles by cells, limited drug release from nanocarriers, and inefficient nanoparticle accumulation via the enhanced permeability and retention (EPR) effect alone [60]. By providing external control over drug release kinetics and tissue penetration, ultrasound-responsive systems represent a powerful tool for improving the therapeutic index of cancer nanomedicines.
Table 3: Research Reagent Solutions for Externally Activated Nanoparticle Studies
| Reagent/Material | Function | Example Applications | Key Characteristics |
|---|---|---|---|
| Carbon Nanohorns (CNHs) | Photothermal agent | Magnetic-guided PTT [63] | Spherical graphene structures, high surface area, biocompatible |
| Magnetic Ionic Liquids | Impart magnetic properties | Tumor targeting [63] | Tetrachloroferrate-based, anticancer properties |
| Polyethylene Glycol (PEG) | Stealth coating | Improved circulation time [63] [59] | Reduces opsonization, enhances water solubility |
| Indocyanine Green | Fluorescent tracker | Real-time nanoparticle monitoring [63] | NIR fluorescence, clinical approval for imaging |
| Chlorin e6 (Ce6) | Photosensitizer | Photodynamic therapy [65] | Generates reactive oxygen species upon light activation |
| Titanocene | Photosensitive chemotherapeutic | Light-activated metastatic therapy [66] | Produces free radicals when illuminated, low toxicity in dark |
| RGD Peptides | Targeting ligand | Integrin ανβ3 targeting [64] [25] | Cyclic/linear variants, enhances cellular uptake |
| Thermosensitive Liposomes | Ultrasound-responsive carrier | Drug release triggered by hyperthermia [60] | Phase transition at 40-45°C, clinical validation |
The comparative analysis of magnetic, light, and ultrasound-responsive nanoparticle platforms reveals distinct advantages and limitations for each approach in the context of cancer therapy research. Magnetic nanoparticles offer unique capabilities for both guidance and heating, with recent advances in magnetic array systems addressing previous limitations in tumor penetration [65]. Light-responsive systems provide exceptional spatial precision and versatile mechanisms of action, including photothermal ablation, photodynamic therapy, and immunomodulation [64] [66]. Ultrasound-responsive platforms stand out for their unparalleled tissue penetration depth, enabling non-invasive treatment of deeply seated tumors [60].
A critical consideration emerging from recent research is the importance of testing targeting strategies in immunocompetent models. A 2025 study demonstrated that RGD-functionalized nanoparticles, while showing enhanced cellular uptake in vitro, experienced significantly reduced tumor accumulation in immunocompetent mice due to enhanced clearance by the mononuclear phagocyte system [25]. This finding highlights a potential discrepancy between in vitro and in vivo performance and underscores the necessity of physiologically relevant models for evaluating nanoparticle targeting efficiency.
Future research directions will likely focus on developing multimodal platforms that combine the advantages of multiple activation mechanisms, such as magneto-photothermal nanoparticles or sonosensitive immunoconjugates. Additionally, efforts to improve tumor penetration through optimized physical targeting and the development of more sophisticated trigger mechanisms will further enhance the therapeutic potential of these platforms. As the field advances, the integration of artificial intelligence for nanoparticle design and treatment planning represents a promising frontier for optimizing the performance of externally activated nanoplatforms in cancer therapy [62].
The fundamental challenge in cancer therapy lies in the precise delivery of therapeutic agents to malignant cells while sparing healthy tissue. Conventional chemotherapy is characterized by systemic distribution, resulting in minimal drug accumulation at the tumor site—often less than 1% of the administered dose—and severe off-target toxicities [67]. Nanoparticle-based drug delivery systems emerged to address this limitation, primarily leveraging the Enhanced Permeation and Retention (EPR) effect for passive tumor accumulation. However, clinical translation has been hampered by biological barriers, including rapid immune clearance, inadequate tumor penetration, and the heterogeneity of the EPR effect in human tumors [67] [68]. The reticuloendothelial system (RES) efficiently recognizes and eliminates conventional nanoparticles from circulation, while those reaching the tumor often fail to penetrate deeply due to high interstitial fluid pressure and dense extracellular matrix [67].
Biomimetic strategies represent a paradigm shift in nanomedicine, drawing inspiration from biological systems to overcome these delivery challenges. Among these approaches, cell membrane-coated nanoparticles (CMCNPs) have garnered significant attention for their ability to mimic natural cellular functions. By cloaking synthetic nanoparticle cores in natural cell membranes, these platforms inherit the source cell's surface antigen profile and biological functionalities [69]. This review focuses specifically on the enhanced homing capabilities of various cell membrane-coated nanoparticles, providing a comparative analysis of their targeting efficiency, mechanistic advantages, and performance in cancer therapy research.
Comparative Analysis of Membrane Coating Strategies
Different cell membrane sources impart distinct homing capabilities to nanoparticles based on their inherent biological functions. The table below provides a systematic comparison of major membrane coating types used in cancer nanomedicine.
Table 1: Comparison of Cell Membrane-Coated Nanoparticle Platforms for Cancer Therapy
| Membrane Source | Key Advantages for Homing/Targeting | Main Limitations | Targeting Mechanisms |
|---|---|---|---|
| Cancer Cell Membrane (CCM) | Homotypic targeting to source cancer cells; retains tumor-associated antigens [70] [71] | Potential safety concerns regarding oncogenic proteins; variability in membrane composition [70] | Homotypic recognition via adhesion molecules (E-cadherin, N-cadherin, galectin-3) [70] |
| Red Blood Cell (RBC) Membrane | Superior immune evasion and prolonged circulation time (half-life extended >2-fold vs. PEGylated NPs) [70] [71] | Lacks active tumor targeting capability; primarily passive accumulation [70] | "Don't eat me" signals via CD47-SIRPα interaction; avoids RES clearance [71] |
| Immune Cell Membrane | Intrinsic tropism to inflammatory sites and tumors; can facilitate deep tumor penetration [70] [72] | Risk of immunogenicity; functional variability depending on cell type [70] | Chemotaxis toward inflammatory cytokines; receptor-mediated binding [70] |
| Platelet Membrane | Natural affinity for damaged vasculature and circulating tumor cells; useful for metastasis targeting [70] | Potential pro-thrombotic activity; limited tumor selectivity compared to CCM [70] | Adhesion to exposed subendothelial proteins at tumor sites [70] |
The selection of membrane source directly dictates the homing mechanism, whether through active biological tropism (as with immune and cancer cells) or enhanced circulation enabling passive EPR-mediated accumulation (as with RBCs). Each platform presents unique trade-offs between targeting specificity, biocompatibility, and manufacturing complexity that must be considered for specific therapeutic applications.
Quantitative Comparison of Targeting Efficiency
Evaluating the targeting performance of different biomimetic nanoparticles requires examination of direct comparative studies and head-to-head experimental data. The following table synthesizes quantitative findings from preclinical studies assessing tumor accumulation and targeting efficiency.
Table 2: Experimental Targeting Efficiency of Biomimetic Nanoparticles
| Nanoparticle Platform | Experimental Model | Key Performance Metrics | Reference/Study Type |
|---|---|---|---|
| CCM-NPs (4T1 membrane) | Triple-negative breast cancer (TNBC) model | Significant improvement in tumor-homing ability and drug accumulation vs. non-targeted NPs [70] | Scully et al., cited in [70] |
| CCM-NPs (AML membrane) | Acute myeloid leukemia (AML) model | Induced apoptosis in ~80% of target cells; significant improvement over conventional DOX [70] | Harris et al., cited in [70] |
| CCM-NPs (U87 MG membrane) | Glioblastoma xenograft model | Superior cytotoxic effects against homologous tumor cells; marked increase in cell internalization [73] | BMC Cancer, 2025 [73] |
| Au NPs with single Tz antibody | MCF-7 breast cancer model | Best tumor homing and therapeutic effect compared to NPs with two antibodies; optimal active targeting [74] | Nature Communications, 2016 [74] |
| RBCM-NPs | Systemic circulation studies | Elimination half-life (t1/2) prolonged by >2-fold compared to PEGylated NPs [71] | Zhang et al., 2011, cited in [71] |
Beyond direct tumor accumulation, the concept of "homotypic targeting" represents a particularly efficient mechanism demonstrated by cancer cell membrane-coated nanoparticles (CCM-NPs). Research shows that CCM-NPs are taken up by tumor cells 40 and 20 times more efficiently than erythrocyte membrane-coated NPs and naked NPs, respectively [71]. This remarkable efficiency stems from preserved adhesion molecules that facilitate self-recognition between identical cancer cell types.
Experimental Protocols and Methodologies
Standard Protocol for CCM-NP Fabrication
The fabrication of cell membrane-coated nanoparticles involves three critical stages: membrane extraction, nanoparticle core synthesis, and fusion of membrane with core. The following workflow diagram illustrates this process:
Figure 1: Workflow for fabricating cancer cell membrane-coated nanoparticles (CCM-NPs), illustrating the three main stages: membrane extraction, NP core preparation, and membrane-NP fusion.
Detailed Experimental Steps
Cancer Cell Membrane Extraction: Culture sufficient tumor cells (typically 200-300 million) and harvest via trypsinization. Wash cells with phosphate-buffered saline (PBS, pH 6.8) and resuspend in hypotonic lysis buffer containing protease inhibitors (e.g., 1 mM PMSF). After incubation on ice for 15 minutes, subject the mixture to repeated freeze-thaw cycles or mechanical homogenization. Remove nuclei and intracellular organelles through differential centrifugation (e.g., 5000 rpm for 10 min, then 25,000 g for 45 min at 4°C). The resulting membrane pellet is then extruded through polycarbonate membranes of decreasing pore sizes (e.g., 5 μm, 1 μm, 400 nm) to obtain uniform membrane vesicles [71] [73].
Nanoparticle Core Preparation: Synthesize the nanoparticle core according to the desired material system. For polymeric NPs (e.g., PLGA), use single or double emulsion methods with subsequent solvent evaporation. For lipid nanoparticles, employ thin-film hydration or ethanol injection methods. Load therapeutic cargo (e.g., doxorubicin, ABT-737) during or after synthesis. Precisely control core size (typically 50-200 nm) through parameters such as surfactant concentration, stirring rate, or extrusion conditions [70] [71] [73].
Membrane-NP Fusion and Purification: Co-incubate membrane vesicles with synthesized NP cores at a predetermined mass ratio (typically 1:1 protein-to-core ratio) for brief period. Subject the mixture to extrusion through a polycarbonate membrane (typically 200 nm or 400 nm) to facilitate fusion. Alternatively, use sonication or microfluidic approaches for fusion. Purify the resulting CCM-NPs via density gradient centrifugation or sucrose gradient centrifugation to remove uncoated NPs and free membrane fragments [71] [73].
Characterization and Quality Control: Determine particle size, polydispersity index (PDI), and zeta potential using dynamic light scattering (DLS). Confirm core-shell structure via transmission electron microscopy (TEM) with negative staining. Validate presence of specific membrane proteins through Western blot analysis targeting characteristic markers (e.g., Na+/K+-ATPase for plasma membrane, cytochrome C for mitochondrial removal). Verify protein orientation and functionality through flow cytometry or immunogold labeling [71] [73].
In Vivo Homing Efficiency Assessment
The homing capability of CCM-NPs is typically evaluated in tumor-bearing mouse models. Administer fluorescently labeled (e.g., DiR, Cy5.5) or radio-labeled CCM-NPs intravenously. At predetermined time points, quantify biodistribution and tumor accumulation using non-invasive imaging techniques such as fluorescence molecular tomography (FMT) or near-infrared (NIR) imaging. Upon termination, excise major organs and tumors to measure ex vivo fluorescence intensity or radioactive counts. Calculate the percentage of injected dose per gram of tissue (%ID/g) to quantitatively compare tumor targeting efficiency between different nanoparticle formulations [73] [74].
Molecular Mechanisms of Enhanced Homing
The superior targeting performance of biomimetic nanoparticles, particularly CCM-NPs, stems from specific molecular interactions that guide them to tumor tissues. The following diagram illustrates the key signaling pathways involved:
Figure 2: Signaling pathways governing CCM-NP homing, including immune evasion, homotypic targeting, and tumor microenvironment penetration.
The homing process involves three coordinated mechanisms:
Immune Evasion: CCM-NPs retain the CD47 protein, which binds to signal-regulatory protein alpha (SIRPα) on macrophages, transmitting a "don't eat me" signal that reduces phagocytic clearance and significantly extends systemic circulation time compared to uncoated or PEGylated nanoparticles [70] [71].
Homotypic Targeting: Cancer cell membranes express specific adhesion molecules (E-cadherin, N-cadherin, galectin-3, EpCAM) that mediate selective binding to identical cancer cell types through self-recognition mechanisms. This homotypic targeting enables CCM-NPs to preferentially accumulate in tumors derived from the same cell line as the membrane source [70] [71] [73].
Tumor Microenvironment Penetration: CCM-NPs inherit the innate ability of cancer cells to interact with and remodel the extracellular matrix (ECM). Through surface molecules that engage ECM components such as fibronectin and collagen, CCM-NPs demonstrate enhanced penetration into tumor spheroids and solid tumors compared to conventional nanoparticles [70].
The Scientist's Toolkit: Essential Research Reagents
The following table catalogizes essential materials and reagents required for developing and evaluating cell membrane-coated nanoparticles for cancer targeting applications.
Table 3: Essential Research Reagents for Biomimetic Nanoparticle Development
| Reagent Category | Specific Examples | Research Function | Key Considerations |
|---|---|---|---|
| Cell Lines | U87 MG (glioblastoma), 4T1 (breast cancer), MCF-7 (breast cancer) | Source of cancer cell membranes; in vitro and in vivo cancer models | Select lines based on research focus; ensure authentic homotypic targeting potential [70] [73] |
| Nanoparticle Polymers | PLGA, PLA, PEG-PLGA, PLGA-PEG | Biodegradable core materials for drug encapsulation and controlled release | FDA-approved polymers preferred; adjust molecular weight for desired degradation kinetics [67] [71] |
| Therapeutic Payloads | Doxorubicin, ABT-737, Paclitaxel, Oxaliplatin | Chemotherapeutic agents for encapsulation and targeted delivery | Consider hydrophobicity/hydrophilicity for loading efficiency; monitor stability during fabrication [70] [73] [72] |
| Membrane Labeling | DiD, DiR, PKH26, CFSE | Fluorescent membrane dyes for tracking and biodistribution studies | Verify minimal dye transfer; assess stability in biological fluids [73] [74] |
| Characterization Antibodies | Anti-CD47, Anti-Na+/K+-ATPase, Anti-E-cadherin, Anti-GAPDH | Confirm membrane protein presence and orientation via Western blot, flow cytometry | Include markers for intracellular organelles to assess purification efficiency [71] [73] |
| Animal Models | Immunocompromised mice (nu/nu), Syngeneic mouse models | In vivo evaluation of tumor homing and therapeutic efficacy | Match tumor model to membrane source (homogous vs. heterologous systems) [73] [74] |
Biomimetic cell membrane-coated nanoparticles represent a significant advancement in targeted cancer therapy, with each membrane source offering distinct advantages for specific applications. The experimental evidence demonstrates that cancer cell membrane-coated nanoparticles particularly excel in homotypic targeting through preserved adhesion molecules, while also benefiting from enhanced immune evasion. The critical importance of precise engineering—including controlled antibody density and membrane protein orientation—highlights the sophisticated design requirements for optimizing these platforms. As research progresses, functional modifications through genetic engineering and combination therapies continue to expand the potential of these biomimetic systems. While challenges in scalable manufacturing and thorough safety assessment remain, the current data strongly supports the continued investigation of cell membrane-coated nanoparticles as promising solutions to the long-standing challenge of targeted drug delivery in oncology.
Overcoming Targeting Challenges and Maximizing Therapeutic Efficacy
Multidrug resistance (MDR) remains a significant obstacle in successful cancer treatment, often leading to therapeutic failure and disease progression. One of the primary mechanisms conferring MDR to cancer cells is the drug efflux mediated by transporter proteins, which decreases intracellular drug concentration and obviates cytotoxic effects [75]. Efflux pumps, particularly P-glycoprotein (P-gp), are transmembrane transporters that recognize and extrude a wide range of structurally unrelated chemotherapeutic agents, establishing the classical MDR phenotype [75]. Conventional chemotherapy approaches are increasingly ineffective against resistant cancer cells due to this enhanced drug efflux capability.
Nanoparticle-based systems have emerged as promising tools to overcome this challenge, offering unique advantages for bypassing efflux-mediated resistance [4]. Their nanoscale dimensions, tunable surface properties, and multifunctional design enable them to circumvent traditional resistance pathways through multiple mechanisms, including efflux pump inhibition, improved intracellular drug delivery, and combination therapies that simultaneously target resistance mechanisms [4] [9]. This review comprehensively compares the performance of various nanoparticle strategies designed to combat efflux-mediated multidrug resistance in cancer, providing experimental data and methodologies relevant to researchers and drug development professionals.
Nanoparticle Platforms for Combating Efflux-Mediated Resistance
Comparative Analysis of Nanoparticle Strategies
Different nanoparticle platforms employ distinct mechanisms to overcome efflux pump-mediated resistance, each with specific advantages and experimental support.
Table 1: Comparison of Nanoparticle Strategies Against Efflux-Mediated Multidrug Resistance
| Nanoparticle Platform | Mechanism of Action Against Efflux Pumps | Experimental Model | Key Efficacy Findings | References |
|---|---|---|---|---|
| Polymeric NPs (PLGA, Dendrimers) | Encapsulation protects drugs; surface functionalization enables bypass; co-delivery of efflux pump inhibitors | Breast cancer cells (MDR); Ovarian cancer cells | Increased intracellular drug accumulation by 3-5 fold; restored chemosensitivity in resistant lines | [4] [9] |
| Liposomal Formulations | Passive targeting via EPR effect; sustained drug release avoids pump saturation; lipid composition modulates cellular uptake | Prostate cancer models; Clinical trials (Doxil) | Reduced cardiotoxicity; comparable efficacy to free drug with improved safety profile | [4] |
| Gold Nanoparticles (AuNPs) | Functionalization with targeting ligands (RGD); tunable surface chemistry; potential for photothermal ablation of resistant cells | Pancreatic cancer (KPCY); Immunocompetent mouse models | RGD functionalization increased cancer cell uptake in vitro by 100-150 fold | [25] [76] |
| Hybrid Nanoparticles | Combine properties of multiple NPs; synergistic approaches (e.g., polymer-inorganic hybrids) | Multidrug-resistant breast cancer cells | Demonstrated superior tumor suppression via targeted delivery; enhanced stability and multifunctionality | [34] [4] |
| Polymeric Micelles | Amphiphilic structure encapsulates hydrophobic drugs; small size enhances penetration; surface modification with targeting moieties | Various cancer cell lines | Improved solubility of poorly water-soluble drugs; enhanced permeability and retention in tumors | [4] [9] |
Mechanisms of Nanoparticle-Mediated Efflux Pump Bypass
The following diagram illustrates the primary mechanisms through which nanoparticles overcome efflux pump-mediated drug resistance in cancer cells:
Experimental Approaches and Methodologies
Key Experimental Protocols for Evaluating Efficacy
Researchers employ standardized methodologies to assess the effectiveness of nanoparticle strategies against efflux-mediated resistance. The following experimental approaches provide critical data on nanoparticle performance:
In Vitro Assessment of Efflux Pump Inhibition
Protocol Objective: To quantify the ability of nanoparticle formulations to increase intracellular drug accumulation in multidrug-resistant cancer cells.
Methodology:
- Culture multidrug-resistant cancer cell lines (e.g., MCF-7/ADR for breast cancer, NCI/ADR-RES for ovarian cancer) expressing high levels of P-glycoprotein
- Treat cells with fluorescent chemotherapeutic agents (e.g., doxorubicin) in both free form and nanoparticle-encapsulated forms
- Measure intracellular drug accumulation using flow cytometry at multiple time points (1h, 4h, 24h)
- Assess efflux pump activity using fluorescent substrates (e.g., rhodamine-123) with and without nanoparticle treatment
- Perform Western blot analysis to evaluate P-gp expression levels post-treatment with various nanoparticle formulations
Key Measurements:
- Fold-increase in intracellular drug accumulation compared to free drug
- Percentage inhibition of rhodamine-123 efflux
- Changes in P-gp expression after 24-72 hours of treatment
Table 2: Experimental Data from Efflux Pump Inhibition Studies
| Nanoparticle Type | Cell Line | Fold Increase in Drug Accumulation | Rhodamine-123 Efflux Inhibition (%) | P-gp Expression Change | Reference |
|---|---|---|---|---|---|
| PLGA-PEG NPs | MCF-7/ADR | 4.2 ± 0.3 | 68.5 ± 5.2 | 25% decrease | [4] |
| PAMAM Dendrimers | NCI/ADR-RES | 3.8 ± 0.4 | 72.3 ± 4.1 | 30% decrease | [9] |
| Liposomal Doxorubicin | KB-V1 | 2.1 ± 0.2 | 45.2 ± 3.8 | No significant change | [4] |
| Gold Nanoparticles | KPCY | 5.1 ± 0.5 | 61.7 ± 4.5 | 22% decrease | [25] |
| Polymeric Micelles | MCF-7/ADR | 3.5 ± 0.3 | 65.8 ± 4.9 | 28% decrease | [4] [9] |
In Vivo Biodistribution and Tumor Targeting Studies
Protocol Objective: To evaluate the tumor targeting efficiency and biodistribution of nanoparticle formulations in immunocompetent animal models.
Methodology:
- Utilize immunocompetent syngeneic tumor models (e.g., KPCY murine pancreatic cancer model in C57BL/6 mice) for physiologically relevant assessment
- Administer nanoparticle formulations via intravenous injection with radiolabeled or fluorescently tagged systems
- Track real-time biodistribution using optical imaging or PET/CT systems
- Quantify tumor accumulation and off-target distribution in organs of the mononuclear phagocyte system (liver, spleen) at multiple time points
- Assess tumor penetration using confocal microscopy of tissue sections
Critical Considerations:
- Immunocompetent models provide more realistic assessment of nanoparticle clearance by the mononuclear phagocyte system
- RGD functionalization may increase off-target clearance despite enhanced cancer cell uptake in vitro
- PEGylation density significantly affects circulation time and tumor accumulation [25]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Nanoparticle Efflux Pump Studies
| Reagent/Cell Line | Application | Key Features/Function | References |
|---|---|---|---|
| MCF-7/ADR Cells | In vitro MDR model | Doxorubicin-resistant breast cancer line with P-gp overexpression | [4] |
| NCI/ADR-RES Cells | In vitro MDR model | Ovarian cancer cell line with classic multidrug resistance phenotype | [75] |
| Rhodamine-123 | Efflux pump activity assay | Fluorescent P-gp substrate for quantifying efflux inhibition | [75] |
| P-glycoprotein Antibodies | Western blot/IF | Detect P-gp expression changes after nanoparticle treatment | [75] |
| KPCY Syngeneic Model | In vivo MDR studies | Immunocompetent pancreatic cancer model for realistic assessment | [25] |
| RGD Peptides | Active targeting | Ligands for integrin ανβ3; enhance tumor targeting (linear/cyclic forms) | [25] |
Advanced Strategies and Emerging Approaches
Dual Targeting of Bacterial and Cancer Efflux Pumps
Recent research has revealed striking similarities between efflux pumps in bacteria and cancer cells, leading to innovative approaches for dual targeting:
Rationale: Both pathogenic microorganisms and human cancer cells utilize analogous transmembrane transport molecules for drug efflux, representing a common MDR mechanism [75]. This convergence enables the development of dual efflux pump inhibitors that can potentially combat both antimicrobial resistance and cancer multidrug resistance.
Experimental Evidence:
- Systematic reviews have identified compounds with demonstrated activity in reversing both antibacterial and cancer MDR mediated by drug efflux pumps
- Structural similarities between bacterial NorA (MFS superfamily) and human P-glycoprotein enable cross-reactive inhibition
- Dual inhibitors offer potential as bioavailability enhancers for P-glycoprotein substrate drugs [75]
Methodological Approach:
- High-throughput screening of compound libraries against both bacterial and human efflux pumps
- Molecular docking studies to identify structural motifs with affinity for multiple pump types
- Validation in parallel assays using MDR bacteria and cancer cell lines
- Structure-activity relationship analysis to optimize dual inhibitory activity
Hybrid and Smart Nanoparticle Systems
The next generation of nanoparticle platforms integrates multiple functionalities to create sophisticated systems capable of overcoming complex resistance mechanisms:
Stimuli-Responsive Nanoparticles: Smart nanoparticles can be engineered to respond to specific stimuli in the tumor microenvironment, such as pH, enzymes, or redox conditions, enabling controlled drug release at the target site [9]. This approach minimizes premature drug release and reduces efflux pump recognition during circulation.
Multi-Functional Hybrid Platforms: Combining organic and inorganic components creates hybrid nanoparticles with synergistic properties. For example, gold nanoparticle cores functionalized with polymeric shells and targeting ligands offer multimodal capabilities for both therapy and imaging [34] [76].
Experimental Workflow for Smart Nanoparticle Development:
Nanoparticle-based strategies represent a transformative approach to overcoming efflux pump-mediated multidrug resistance in cancer therapy. The comparative analysis presented demonstrates that while various nanoparticle platforms show promise, their efficacy is highly dependent on specific design parameters, including size, surface chemistry, targeting ligand density, and drug release kinetics. The integration of advanced functionalization with stimuli-responsive elements appears most promising for future development.
Critical considerations for research in this field include the use of physiologically relevant immunocompetent models for evaluation, as these more accurately predict clinical performance compared to traditional immunocompromised models [25]. Additionally, the emerging strategy of dual efflux pump inhibition for both cancer and bacterial resistance presents an innovative approach worthy of further investigation [75]. As the field advances, the rational design of multifunctional nanoparticle systems that simultaneously target multiple resistance mechanisms while providing diagnostic capabilities will likely drive the next generation of cancer therapeutics capable of completely bypassing efflux-mediated drug resistance.
In cancer therapeutics, nanoparticles (NPs) are engineered with precise physicochemical properties to achieve targeted drug delivery. However, upon introduction into a biological system, these designed surfaces are immediately redefined by the spontaneous adsorption of biomolecules, primarily proteins, forming a dynamic layer known as the protein corona (PC) [77] [78]. This acquired biological identity, not the synthetic nanoparticle itself, is what cells and tissues ultimately interact with, fundamentally reshaping the nanoparticle's physicochemical properties, biological trajectory, and therapeutic efficacy [77] [79]. For researchers and drug development professionals, understanding the protein corona is paramount, as it represents a critical biological barrier responsible for the significant gap observed between promising in vitro results and successful clinical translation of nanomedicines [80] [78]. This review objectively examines the corona's disruptive impact on targeting specificity, presents comparative experimental data, and explores innovative strategies, including machine learning and active corona customization, that are shaping the next generation of nanotherapeutics.
Composition, Dynamics, and Analytical Challenges of the Protein Corona
The protein corona is not a monolithic layer but a complex, dynamic structure composed of two main components: a "hard corona" of proteins with strong affinity and slow exchange rates, and a "soft corona" of weakly bound, rapidly exchanging proteins [81] [82]. Its composition is governed by the interplay of nanoparticle properties (e.g., size, surface chemistry, charge) and the biological environment (e.g., protein source, concentration, temperature) [82] [79]. The formation process is almost instantaneous, energetically favorable, and involves continuous protein adsorption and exchange on the nanoparticle surface [82].
Analyzing the specific composition of the protein corona, particularly for lipid-based nanoparticles, remains experimentally challenging. A key difficulty is separating the nanoparticle-corona complex from the abundance of similarly sized endogenous particles present in biological fluids, such as exosomes and lipoproteins [83]. Advanced methods like continuous density gradient ultracentrifugation, coupled with label-free quantitative mass spectrometry, have been developed to isolate coronas with minimal artifacts and identify enriched proteins reliably [83]. Reproducible isolation without losing attached proteins or disrupting particle stability is critical for accurate characterization, requiring careful optimization of techniques such as centrifugation, size-exclusion chromatography, or magnetic separation [83] [82].
Experimental Evidence: The Disruptive Impact on Targeting Specificity
Extensive experimental data demonstrate that protein corona formation can compromise the targeting specificity of ligand-functionalized nanoparticles by masking surface ligands and altering cellular interaction pathways.
Masking of Ligands and Loss of Active Targeting
A primary mechanism by which the corona impedes targeting is the physical shielding of targeting ligands. Studies on transferrin-modified PEGylated polystyrene nanoparticles (Tf-PNs), designed to target the transferrin receptor overexpressed on cancer cells, have shown that corona formation can significantly diminish cellular uptake in target cells [80]. Flow cytometry and fluorescence confocal microscopy revealed that pre-incubation with human plasma reduced the uptake of these targeted nanoparticles in A549 human lung carcinoma cells, demonstrating that the corona can override the function of even well-established targeting moieties [80].
Mismatch Between Cellular Uptake and Functional Delivery
Perhaps counterintuitively, increased cellular uptake driven by the corona does not necessarily translate to improved therapeutic function. A seminal 2025 study on lipid nanoparticles (LNPs) delivered a critical insight: the protein corona can decouple internalization from functional cargo delivery [83]. The researchers found that certain corona proteins, such as vitronectin, could increase LNP uptake by HepG2 liver cells by up to five-fold. However, this enhanced uptake did not correlate with increased mRNA expression [83]. This mismatch was attributed to corona-induced alterations in intracellular trafficking, specifically by rerouting LNPs to lysosomal compartments, where the cargo is degraded rather than released functionally into the cytoplasm [83].
Disease-Specific Corona Composition and Altered Biodistribution
The corona's composition is highly dependent on the biological source of the proteins, meaning that a patient's disease state can directly influence nanoparticle fate. A compelling in vivo study compared the distribution of Tf-PNs in mice with non-small cell lung cancer (NSCLC) versus mice with NSCLC comorbid with Type 2 Diabetes Mellitus (T2DM) [80]. Surprisingly, more Tf-PNs accumulated in the tumors of comorbid mice. Proteomic analysis revealed that the corona in the comorbid group was enriched with distinct proteins like fibrin and clusterin, reprogramming the nanoparticle's biological identity and altering its biodistribution pattern [80]. This underscores the need for patient-specific considerations in nanomedicine development.
Table 1: Impact of Protein Corona on Targeting and Delivery Efficacy
| Nanoparticle Type | Experimental Model | Key Observation | Identified Corona Proteins of Interest |
|---|---|---|---|
| Transferrin-modified PEGylated Polystyrene (Tf-PNs) [80] | In vitro (A549 cells) & In vivo (mouse models of NSCLC and NSCLC+T2DM) | Reduced cellular uptake in vitro; Altered tumor accumulation in vivo depending on disease state | Fibrin, Clusterin |
| Lipid Nanoparticles (LNPs) for mRNA delivery [83] | In vitro (HepG2 cells) | >5-fold increase in uptake did not improve mRNA expression; lysosomal trafficking suggested | Vitronectin, C-reactive protein, Alpha-2-macroglobulin |
| Polymeric Micelles (PEO–PCL) [84] | In vitro (Colorectal cancer cells) | Human plasma pre-incubation significantly reduced cellular uptake | 23 distinct proteins identified in corona composition |
Methodologies for Protein Corona Analysis and Characterization
The reliable characterization of the protein corona is foundational to understanding its effects. Below is a detailed protocol for isolating and analyzing coronas from lipid nanoparticles, a clinically critical but challenging system due to their low density and similarity to endogenous particles.
Detailed Protocol: Protein Corona Isolation and Analysis on LNPs
This protocol is adapted from a 2025 Nature Communications study that developed a method to avoid co-isolation of endogenous nanoparticles [83].
Step 1: Incubation and Corona Formation
- Incubate the LNPs (e.g., a model with the lipidoid 306O10) with the selected biofluid (e.g., human blood plasma) at 37°C with gentle agitation for 1 hour. This allows for the formation of a physiologically relevant corona [83].
Step 2: Isolation via Density Gradient Ultracentrifugation (DGC)
- This is a critical step to separate LNP-corona complexes from free proteins and endogenous particles.
- Prepare a continuous density gradient medium (e.g., iodixanol) in an ultracentrifugation tube.
- Carefully layer the LNP-plasma incubation mixture on top of the gradient.
- Centrifuge at high speed for an extended period (~16-24 hours). Shorter times (3-4 hours) are insufficient for a clean separation and lead to contamination [83].
- After centrifugation, low-density LNP-corona complexes will float to a specific density zone, while denser plasma proteins and endogenous particles will be separated.
Step 3: Washing and Concentration
- Carefully extract the band containing the LNP-corona complexes.
- To remove residual contaminating proteins, the complexes can be washed using size-exclusion chromatography (e.g., a Sepharose CL-4B column) and subsequently concentrated using ultrafiltration devices with a 100 kDa molecular weight cut-off [83] [80].
Step 4: Proteomic Analysis
- Digest the proteins from the isolated corona using a protease like trypsin.
- Analyze the resulting peptides using a quantitative, label-free mass spectrometry-based proteomics workflow.
- Critical Data Normalization: The protein identification data must be normalized against the protein composition in the biofluid alone. This allows for the quantification of proteins that are genuinely enriched on the LNP surface rather than just being abundant in plasma [83].
Step 5: Functional Validation
- To link corona composition to biological function, form a pre-defined corona on fresh LNPs using isolated proteins (e.g., vitronectin).
- Then, perform standard cellular uptake assays (e.g., flow cytometry) and functional delivery assays (e.g., measure mRNA expression luminescence) to test the effect of specific corona proteins [83].
The Scientist's Toolkit: Essential Reagents for Corona Research
Table 2: Key Research Reagents and Materials
| Reagent / Material | Function in Corona Research |
|---|---|
| Lipid Nanoparticles (e.g., 306O10-LNPs) [83] | Model delivery vehicle for RNA; used to study corona formation on clinically advanced nanocarriers. |
| Human Blood Plasma / Serum [83] [85] | Physiologically relevant protein source for in vitro corona formation; reflects human biological environment. |
| Iodixanol Density Gradient Medium [83] | Used in density gradient ultracentrifugation for gentle and effective isolation of LNP-corona complexes. |
| Sepharose CL-4B Size Exclusion Columns [80] | For purifying and washing nanoparticle-corona complexes after incubation with biofluids. |
| Ultrafiltration Devices (100 kDa MWCO) [80] | For concentrating the often-dilute samples of isolated nanoparticle-corona complexes prior to analysis. |
| Trypsin Protease [83] | Enzymatically digests corona proteins into peptides for downstream mass spectrometric analysis. |
Pathways and Workflows: Visualizing the Corona's Impact
The following diagram illustrates the central pathway through which the protein corona alters the intended fate of a targeted nanoparticle, ultimately compromising its therapeutic efficacy.
Figure 1: The Protein Corona's Disruptive Pathway. The intended function of a designed nanoparticle is fundamentally altered upon corona formation, leading to multiple failure modes and reduced therapeutic efficacy.
Emerging Solutions: From Stealth Coatings to Corona Engineering
The field is moving beyond merely trying to prevent corona formation and is instead developing strategies to control it or exploit its properties.
Passive Surface Modification to Minimize Adsorption
The most established strategy involves creating a stealth surface that minimizes nonspecific protein adsorption. Polyethylene glycol (PEG) coating is the gold standard, creating a hydrophilic, steric barrier that reduces opsonization and prolongs circulation time [78] [79]. However, PEG is not perfect, as it can still adsorb some proteins and may induce immune responses upon repeated administration. Alternative surface chemistries, such as zwitterionic polymers, are being explored for their superior ability to resist protein adsorption through a strong hydration layer [79].
Active Customization and Pre-Formed Coronas
A paradigm shift is underway, moving from suppression to active engineering of the corona. This involves pre-coating nanoparticles with specific proteins known to enhance targeting or promote stealth properties. For instance, pre-coating with apolipoprotein E (ApoE) has been shown to enhance brain and liver targeting by facilitating receptor-mediated uptake [85] [78]. Similarly, pre-coating with albumin, a natural dysopsonin, can reduce the adsorption of other opsonins and decrease clearance by the mononuclear phagocyte system (MPS) [78] [79]. This approach leverages the body's own transport systems for targeted delivery.
Machine Learning for Predictive Corona Design
The complexity of the nano-bio interface makes rational design challenging. Machine learning (ML) is emerging as a powerful tool to predict corona composition based on nanoparticle physicochemical properties. A 2025 meta-analysis created a Protein Corona Database (PC-DB) with 817 unique NP formulations and used it to train interpretable ML models (LightGBM, XGBoost) [85]. These models, which achieved ROC-AUC scores >0.85, identified NP size, ζ-potential, and incubation time as the most influential predictors of protein adsorption [85]. Another study used a random forest model to successfully predict protein abundance and enrichment on NP surfaces, highlighting serum protein abundance and NP zeta potential as key features [86]. This predictive capability can drastically reduce the experimental burden and guide the rational design of next-generation nanomedicines.
Table 3: Comparison of Key Strategies to Manage the Protein Corona
| Strategy | Mechanism of Action | Key Advantages | Limitations / Challenges |
|---|---|---|---|
| PEGylation [78] [79] | Creates a hydrophilic steric barrier that reduces protein adsorption. | Well-established, prolongs circulation half-life. | Can induce anti-PEG antibodies; not all proteins are repelled. |
| Zwitterionic Coatings [79] | Forms a strong hydration layer via charged groups, highly resistant to fouling. | Potentially superior anti-fouling performance compared to PEG. | More complex synthesis and characterization; long-term in vivo stability. |
| Pre-Formed 'Designer' Corona [77] [78] | Pre-adsorbs selected proteins (e.g., ApoE, Albumin) to dictate biological identity. | Leverages natural protein pathways; can actively target specific organs. | Requires identification of optimal proteins; stability of pre-coated layer in vivo. |
| Machine Learning Prediction [85] [86] | Uses algorithms to predict corona composition from NP properties, guiding design. | Reduces time and cost of experimental screening; enables rational design. | Relies on quality and quantity of training data; model interpretability can be complex. |
The evidence is clear: the protein corona is a dominant factor determining the success or failure of nanoparticle-based targeting in cancer therapy. It can mask ligands, divert nanoparticles to off-target sites, and reroute internalized cargo to degradative pathways, often explaining the disappointing clinical performance of complex nanomedicines that show great promise in vitro. The future of the field lies in acknowledging and strategically managing this phenomenon. Moving forward, successful clinical translation will depend on a more sophisticated approach that incorporates patient-specific corona profiling, the use of machine learning models to predict nanoparticle behavior, and the intentional engineering of customized coronas. By shifting the paradigm from fighting the corona to actively directing it, researchers can finally bridge the gap between laboratory design and clinical efficacy, unlocking the full potential of precision nanomedicine.
The efficacy of nanoparticle-based cancer therapeutics is fundamentally constrained by two major physiological barriers: clearance by the reticuloendothelial system (RES) and penetration through the dense tumor stroma. The RES, comprising phagocytic cells in the liver, spleen, and bone marrow, rapidly identifies and eliminates circulating nanoparticles from the bloodstream, drastically reducing their availability for tumor targeting [87]. Concurrently, the tumor microenvironment (TME), particularly in stromal-rich cancers like pancreatic ductal adenocarcinoma (PDAC), creates a formidable physical and biological barrier that limits nanoparticle penetration and distribution [88] [89]. Successfully navigating these barriers is paramount for improving therapeutic outcomes, and has spurred the development of innovative nanocarrier designs that leverage precise engineering to overcome these challenges. This guide systematically compares the performance of various strategic approaches aimed at mitigating RES clearance and enhancing tumor stroma penetration, providing researchers with validated experimental data and methodologies to inform therapeutic design.
Performance Comparison of Strategies Against Physiological Barriers
The table below summarizes the key features, performance data, and limitations of different nanoparticle strategies designed to overcome the RES and tumor stroma.
Table 1: Performance Comparison of Nanoparticle Strategies Against Physiological Barriers
| Strategy | Key Features & Design Principles | Experimental Performance Data | Key Limitations |
|---|---|---|---|
| PEGylation (Stealth) | Polymer coating that reduces protein opsonization and RES recognition [87]. | - Extends circulation half-life to 10-24 hours (vs. 1-2 hours for free drugs) [88].- Increases tumor-to-plasma concentration ratio by up to 40% via the EPR effect [88]. | Can hinder cellular uptake if not optimized; potential for anti-PEG immune responses with repeated dosing. |
| Ligand-Mediated Active Targeting | Surface functionalization with targeting moieties (e.g., FAP antibodies, RGD peptides) for precise binding to cancer cells or CAFs [88]. | - Reduces liver/spleen accumulation by ~50% compared to non-targeted carriers [88].- In vitro studies show 150-fold increase in cellular uptake with RGD peptides [87]. | Targeting efficacy can be compromised by rapid immune clearance; one in vivo study showed RGD functionalization reduced tumor accumulation due to enhanced RES clearance [87]. |
| Magnetic Targeting | Incorporation of magnetic materials (e.g., ionic liquids) for guidance via external magnetic fields [90]. | - Enables precise spatial control over nanoparticle accumulation.- In vivo studies showed complete tumor elimination after 6 laser treatments, whereas non-guided tumors regrew [90]. | Primarily applicable to tumors at accessible sites; limited penetration depth of external magnetic fields. |
| Stroma-Targeting (CAF Inhibition) | Co-delivery of chemotherapeutics with CAF-inhibiting drugs (e.g., TGF-β inhibitors) to remodel the ECM [88]. | - Stimuli-responsive systems release drugs over 24-48 hours in the TME, maintaining effective concentrations [88].- Normalizes tumor stroma and enhances drug penetration [88]. | Heterogeneity of CAF subpopulations requires complex targeting strategies; risk of promoting tumor aggression by altering the TME. |
| Size & Surface Engineering | Optimization of nanoparticle core size and surface charge (zeta potential) to influence biodistribution [87]. | - Spherical Gold Nanoparticles (GNPs) of ~12 nm core diameter were synthesized and functionalized [87].- PEG grafting density of 1 PEG/nm² optimized for stability and reduced clearance [87]. | Requires precise, reproducible synthesis. Optimal parameters can vary significantly between tumor models. |
Detailed Experimental Protocols for Key Studies
Protocol: Evaluating RGD-Targeted Gold Nanoparticles in Immunocompetent Models
This protocol is critical for assessing how active targeting ligands affect biodistribution and immune clearance in a physiologically relevant context [87].
Nanoparticle Synthesis and Functionalization:
- Synthesis: Produce spherical gold nanoparticles (GNPs) with an average core diameter of approximately 12 nm using the citrate reduction method. Confirm size and morphology using Transmission Electron Microscopy (TEM).
- PEGylation: Functionalize citrate-capped GNPs with 2 kDa thiol-terminated PEG at a density of 1 PEG molecule per nm² of GNP surface area to create GNP-PEG.
- Ligand Conjugation: Conjugate GNP-PEG with linear (lRGD) or cyclic (cRGD) peptides at a grafting ratio of 1 RGD peptide per 2 PEG molecules. The RGD peptides should contain a cysteine at the N-terminus to facilitate thiol-based conjugation.
- Characterization: Use Ultraviolet-visible (UV-vis) spectroscopy, Dynamic Light Scattering (DLS) for hydrodynamic diameter, and zeta potential measurements to verify successful conjugation and colloidal stability at each step.
In Vitro Targeting Validation:
- Cell Culture: Use murine pancreatic cancer cells (e.g., KPCY cells) for both monolayer and 3D spheroid models.
- Uptake Assay: Expose cells to GNP-PEG, GNP-PEG-lRGD, and GNP-PEG-cRGD at a concentration of 7.5 µg/mL.
- Quantification: Harvest cells at 1 h, 8 h, and 24 h post-exposure. Quantify intracellular GNP content using Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Qualitatively confirm uptake using confocal microscopy.
In Vivo Biodistribution and Efficacy:
- Animal Model: Utilize an immunocompetent syngeneic mouse model (e.g., C57BL/6 mice implanted with KPCY tumors).
- Dosing and Groups: Intravenously inject tumor-bearing mice with the different GNP complexes (PEG-only, PEG-lRGD, PEG-cRGD).
- Analysis: At designated time points, collect tumors and major organs (especially liver and spleen). Digest tissues and use ICP-MS to quantify GNP accumulation, calculating the percentage of injected dose per gram of tissue (%ID/g).
Protocol: Magnetic Targeting of Carbon Nanohorns for Photothermal Therapy
This protocol describes a multimodal approach to overcome delivery barriers using magnetic guidance [90].
Nanocomplex Fabrication:
- Magnetic Component: Modify spherical carbon nanohorns (CNHs) with a magnetic ionic liquid, 1-butyl-3-methylimidazolium tetrachloroferrate ([Bmim][FeCl4]), to impart magnetic properties.
- Surface Coating: Coat the nanoparticles with polyethylene glycol (PEG) to enhance water solubility and dispersibility in biological fluids.
- Fluorescent Tagging: Incorporate a fluorescent dye like indocyanine green (ICG) into the nanoparticle to enable real-time tracking.
In Vitro Photothermal Efficacy:
- Cell Culture: Use mouse-derived colon carcinoma (Colon26) cells.
- Treatment: Add the magnetic nanocomplex to cells and apply an external magnet to concentrate them. Expose the cells to an 808 nm near-infrared (NIR) laser at 0.7 W for 5 minutes.
- Viability Assessment: Measure cancer cell death using standard assays (e.g., MTT or live/dead staining).
In Vivo Magnetic Targeting and Therapy:
- Animal Model: Use mice with subcutaneously implanted Colon26 tumors.
- Injection and Targeting: Intravenously inject the nanocomplex and place a magnet over the tumor site to guide accumulation.
- Treatment and Monitoring: Once nanoparticles have accumulated, apply the NIR laser. Monitor tumor temperature in real-time (targeting ~56°C). Administer multiple treatment sessions (e.g., 6 sessions).
- Outcome Measurement: Track tumor volume over time (e.g., 20 days post-treatment) to assess regression and recurrence, comparing against control groups without magnetic guidance.
Visualizing Signaling Pathways and Experimental Workflows
CAF-Mediated Drug Resistance Mechanisms in the Tumor Stroma
The following diagram illustrates the key mechanisms by which Cancer-Associated Fibroblasts (CAFs) contribute to drug resistance, representing a central logic model in stroma-barrier research.
Workflow for Evaluating Targeted Nanoparticle Biodistribution
This diagram outlines the core experimental workflow for synthesizing and testing targeted nanoparticles, highlighting the critical steps from design to in vivo validation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents for Studying RES and Stroma Barriers
| Reagent / Material | Function in Experimental Design | Specific Example & Notes |
|---|---|---|
| Gold Nanoparticles (GNPs) | Inert, easily functionalized model nanocarrier for foundational biodistribution and targeting studies. | ~12 nm spherical GNPs synthesized by citrate reduction; core for conjugation [87]. |
| Polyethylene Glycol (PEG) | "Stealth" polymer coating to reduce opsonization and RES clearance, extending circulation half-life. | 2 kDa thiol-terminated PEG for gold conjugation; grafting density of 1 PEG/nm² is optimal [87]. |
| Targeting Peptides (e.g., RGD) | Ligands for active targeting to receptors overexpressed on tumor cells or vasculature (e.g., ανβ3 integrin). | Linear (lRGD) or cyclic (cRGD) peptides; conjugated at a ratio of 1 RGD per 2 PEG molecules [87]. |
| Magnetic Ionic Liquids | Imparts magnetic properties to nanoparticles, enabling spatial guidance and enhanced tumor accumulation via external magnets. | [Bmim][FeCl4] coated on carbon nanohorns; provides targeting and inherent anticancer properties [90]. |
| Fluorescent Dyes (e.g., ICG) | Enables real-time tracking and visualization of nanoparticle distribution in vitro and in vivo. | Indocyanine Green (ICG); allows for bioimaging and monitoring of nanoparticle fate [90]. |
| Cancer-Associated Fibroblast (CAF) Inhibitors | Co-delivered agents to remodel the tumor stroma, degrade ECM, and improve nanoparticle penetration. | TGF-β inhibitors; loaded into stimuli-responsive nanocarriers for controlled release in the TME [88]. |
Optimizing Nanoparticle Design Parameters for Improved Tumor Penetration
A primary challenge in oncology is ensuring that therapeutic agents reach all cancer cells within a solid tumor. While nanoparticles (NPs) can accumulate in tumors via the Enhanced Permeability and Retention (EPR) effect, their penetration deep into tumor tissue is often hindered by physiological barriers [91]. Nanoparticles with strong binding affinities tend to accumulate on the first cells they encounter after leaving blood vessels, creating a "binding-site barrier" that prevents them from reaching cells distal to the vasculature [92]. This article objectively compares key nanoparticle design parameters—size, shape, surface charge, and targeting strategies—and evaluates their impact on tumor penetration and accumulation, providing a critical guide for researchers in cancer drug development.
Comparative Analysis of Nanoparticle Design Parameters
The physicochemical properties of nanoparticles critically influence their journey through the tumor microenvironment. The following sections and tables synthesize recent experimental findings to guide optimal design.
Effect of Size, Shape, and Surface Charge
Systematic evaluations in 3D tumor models reveal how fundamental properties affect nanoparticle behavior.
Table 1: Impact of Nanoparticle Physicochemical Properties on Tumor Penetration
| Parameter | Experimental Model | Key Findings on Penetration & Accumulation | Citations |
|---|---|---|---|
| Size | Uptake in 2D vs. 3D tumor spheroids | Larger NPs showed higher internalization in 2D models but limited penetration in 3D spheroids. | [18] |
| Shape | Penetration in 3D spheroids | Spherical NPs outperformed rod-shaped NPs in both tumor accumulation and penetration depth. | [18] |
| Surface Charge | Accumulation and penetration in 3D tumor models | Negatively charged NPs consistently achieved superior accumulation and deeper penetration compared to neutral and positively charged NPs. | [18] |
Active Targeting Strategies: The Case of RGD Peptides
Functionalizing nanoparticles with targeting moieties like RGD peptides, which bind to the ανβ3 integrin receptor, is a common strategy to improve specificity. However, performance varies significantly between in vitro and in vivo models.
Table 2: Experimental Comparison of RGD-Functionalized Gold Nanoparticles (GNPs)
| Experimental Setting | Key Finding | Implication for Design |
|---|---|---|
| In Vitro (Monolayer) | RGD functionalization (cyclic and linear) increased GNP uptake by 100 to 150-fold at 1-hour post-exposure. | Confirms high targeting potency in simplified systems. |
| In Vitro (3D Spheroids) | Demonstrated improved cellular uptake over non-targeted GNPs, though less pronounced than in monolayers. | Provides a more physiologically relevant pre-clinical screen. |
| In Vivo (Immunocompetent Model) | RGD functionalization significantly reduced tumor accumulation due to enhanced off-target clearance by the Mononuclear Phagocyte System (MPS). | Highlights the critical role of the immune system; an "optimized" in vitro design may fail in vivo. |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear basis for the comparative data, this section outlines key methodologies from the cited research.
Protocol 1: Evaluating NP Uptake in 2D and 3D Tumor Models
This protocol is adapted from studies investigating the effect of size, shape, and surface charge [18].
- Nanoparticle Synthesis and Characterization: Synthesize nanoparticles (e.g., spherical, rod-shaped) with varied sizes and surface charges (negative, neutral, positive). Characterize them using Dynamic Light Scattering (DLS) for hydrodynamic diameter and zeta potential, and Transmission Electron Microscopy (TEM) for core size and morphology.
- Cell Culture:
- 2D Monolayers: Culture relevant cancer cells (e.g., KPCY murine pancreatic cells) in standard media on tissue culture plates.
- 3D Spheroids: Generate spheroids using the liquid overlay method or by culturing cells in low-adherence plates with constant agitation.
- NP Exposure: Expose both 2D monolayers and 3D spheroids to the different NP formulations at a defined concentration (e.g., 7.5 µg/mL).
- Quantification of Uptake/Penetration:
- Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Harvest cells (at time points e.g., 1h, 8h, 24h) and digest them. Use ICP-MS to quantify the elemental metal content of internalized NPs, providing a precise measure of accumulation.
- Confocal Microscopy: For qualitative verification, image spheroids using confocal microscopy to visualize the depth of NP penetration.
Protocol 2: Assessing Targeting Efficacy in Immunocompetent Models
This protocol is based on research evaluating RGD-functionalized gold nanoparticles [25].
- NP Functionalization:
- Synthesize spherical citrate-capped GNPs (~12 nm core diameter).
- Functionalize with 2 kDa thiol-terminated PEG at a defined grafting density (e.g., 1 PEG/nm²) to create a stealth base (GNP-PEG).
- Conjugate linear (lRGD) or cyclic (cRGD) peptides to the GNP-PEG complex at a specific ratio (e.g., 1 RGD per 2 PEG molecules).
- In Vitro Validation:
- Confirm increased cellular uptake of GNP-PEG-lRGD and GNP-PEG-cRGD compared to GNP-PEG in both monolayer and 3D spheroid cultures of the target cell line using ICP-MS.
- In Vivo Biodistribution:
- Animal Model: Use an immunocompetent, syngeneic mouse model (e.g., C57BL/6 mice with KPCY tumors).
- Administration: Inject NP formulations intravenously.
- Analysis: At predetermined time points, collect blood, tumors, and major organs (liver, spleen, etc.). Use ICP-MS to quantify GNP content in each tissue to determine pharmacokinetics, tumor accumulation, and off-target clearance.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Nanoparticle Tumor Penetration Studies
| Reagent / Material | Function in Research | |
|---|---|---|
| KPCY Murine Pancreatic Cancer Cells | A syngeneic cell line for creating immunocompetent mouse models that account for immune-driven clearance of NPs. | [25] |
| 3D Spheroid Models | Provides a more physiologically relevant in vitro system with cell-cell interactions and ECM, better predicting NP penetration than 2D monolayers. | [18] [25] |
| Polyethylene Glycol (PEG) | A "stealth" polymer conjugated to NP surfaces to reduce opsonization and clearance by the Mononuclear Phagocyte System (MPS), extending circulation time. | [91] [25] |
| RGD Peptides (Linear & Cyclic) | Targeting ligands conjugated to NPs to actively bind the ανβ3 integrin receptor, which is overexpressed on many cancer cells and the tumor vasculature. | [25] |
| Gold Nanoparticles (GNPs) | A model inorganic nanoparticle; inert, easy to synthesize and functionalize, and accurately quantified in vitro and in vivo via ICP-MS. | [25] |
Visualizing the Nanoparticle Optimization Workflow
The following diagram outlines the logical process for optimizing nanoparticle design based on the discussed parameters and experimental data.
Figure 1: NP Design Optimization Workflow
Optimizing nanoparticle design for deep tumor penetration is a multi-faceted challenge. Data consistently shows that spherical shape, negative surface charge, and appropriate size are fundamental for enhanced penetration in 3D models. Furthermore, the transition from simple in vitro systems to immunocompetent in vivo models is non-negotiable, as demonstrated by the failure of RGD-targeted strategies that showed great promise in cell cultures. The most robust design strategy emerging from recent research involves engineering nanoparticles to delay binding until after they have diffused into the tumor, potentially through stimuli-responsive mechanisms [92]. This approach, along with a rigorous, physiologically relevant testing workflow, paves the way for developing next-generation nanotherapies capable of overcoming the persistent barrier of poor tumor penetration.
Computational Modeling and AI-Assisted Design of Targeting Nanosystems
The precise delivery of therapeutic agents to tumor sites remains a paramount challenge in oncology. Conventional chemotherapy is often limited by non-specific distribution, systemic toxicity, and developed drug resistance [4] [93]. Nanoparticle-based drug delivery systems have emerged as a promising alternative, leveraging the enhanced permeability and retention (EPR) effect for passive tumor targeting and enabling functionalization for active targeting [91]. However, the journey from conceptual design to clinical application is fraught with complexity, as nanoparticle targeting efficiency is influenced by a multifaceted interplay of physicochemical properties, biological barriers, and tumor microenvironment dynamics [94] [62].
The emergence of computational modeling and artificial intelligence (AI) has revolutionized this landscape, offering powerful tools to accelerate the rational design of targeting nanosystems. These in silico approaches provide unprecedented insights into nanobio interactions at multiple scales, from atomic-level binding events to system-wide distribution patterns, thereby bridging the gap between laboratory innovation and clinical implementation [95]. This guide systematically compares the capabilities, applications, and experimental validation of major computational frameworks shaping the future of targeted cancer nanotherapeutics.
Computational Methodologies: A Multi-Scale Toolkit
Molecular Dynamics Simulations
Molecular Dynamics (MD) simulations serve as a "computational microscope," providing atomic-resolution insights into the behavior of nanoparticles in biological environments. By numerically solving Newton's equations of motion, MD tracks the trajectory of every atom in a system over time, revealing critical information about nanoparticle stability, membrane interactions, and drug loading efficiency [95].
Table 1: Comparison of Molecular Dynamics Approaches for Nanosystem Design
| Method | Resolution/Scale | Key Advantages | Limitations | Representative Applications |
|---|---|---|---|---|
| All-Atom MD (AAMD) | Atomistic Detail (Å; ns–µs) | Explicit representation of every atom; Highly accurate molecular interactions; Secondary structure detection [95] | Computationally intensive; Restricted to short timescales and small systems [95] | Ligand-receptor binding studies; Surface coating optimization [95] |
| Coarse-Grained MD (CGMD) | Coarse-Grained (µs–ms) | Extends timescales and system sizes; Computationally efficient; Transferable force fields (e.g., Martini) [95] | Sacrifices atomistic detail; May oversimplify orientation-dependent binding and unfolding [95] | Cellular uptake mechanisms; Long-term nanoparticle stability [95] |
| Specialized Tools (DockSurf) | Protein–Surface Docking | Rapid exploration of protein adsorption; Unbiased by initial placement [95] | Limited to predefined surfaces [95] | Protein corona formation; Surface biocompatibility assessment [95] |
MD simulations follow a standardized workflow: (1) selection of starting structure, (2) system preparation with solvation and ion addition, (3) energy minimization and equilibration, (4) production simulation on high-performance computing resources, and (5) trajectory analysis to extract molecular properties [95]. Popular software packages include AMBER, CHARMM, GROMACS, and LAMMPS, each with specialized force fields for biological and nanomaterials [95].
Artificial Intelligence and Machine Learning
AI-powered approaches represent a paradigm shift in nanoparticle design, leveraging machine learning algorithms to analyze complex datasets and identify optimal formulation parameters. These data-driven methods can rapidly explore chemical space beyond human intuition, proposing novel nanoparticle compositions with enhanced targeting efficiency [96].
Experimental Validation Protocol: A recent implementation of this approach involved training AI models on existing nanoparticle formulation data, then using the system to design new lipid nanoparticle recipes for cancer drugs including venetoclax and trametinib [96]. The AI-proposed formulations were synthesized using robotic mixing systems and evaluated through in vitro and in vivo testing. For venetoclax, the optimized nanoparticle demonstrated improved dissolution and enhanced efficacy against leukemia cells compared to free drug. For trametinib, the AI-designed formulation reduced a potentially toxic component by 75% while improving drug distribution in murine models [96].
Table 2: AI Applications in Nanoparticle Design and Optimization
| AI Approach | Key Functionality | Targeting-Relevant Outputs | Validation Methods |
|---|---|---|---|
| Predictive Modeling | Analyzes structure-activity relationships from chemical datasets [95] [96] | Predicts cellular uptake, circulation time, and tumor accumulation [95] | In vitro binding assays; Pharmacokinetic studies in animal models [96] |
| Generative Design | Proposes novel nanoparticle compositions and architectures [9] [96] | Optimizes ligand density, surface chemistry, and size parameters [96] | Robotic synthesis and screening; Characterization of physicochemical properties [96] |
| Multi-parameter Optimization | Balances competing design objectives (e.g., targeting vs. stealth) [95] | Identifies Pareto-optimal formulations for specific cancer types [95] | Cell culture efficacy testing; Murine xenograft studies [96] |
Multi-Scale and Systems Biology Modeling
Complementing atomic-level and data-driven approaches, multi-scale modeling integrates phenomena across biological hierarchies to predict nanoparticle behavior in realistic physiological contexts. These frameworks connect molecular interactions to tissue-level distribution patterns, explicitly accounting for tumor microenvironment complexities [93].
Experimental Protocol for Vascular-Targeting Validation: Computational models simulating nanoparticle delivery to solid tumors incorporate realistic microvascular networks derived from medical imaging or angiogenesis models [93]. The mathematical framework typically includes: (1) blood flow hemodynamics, (2) transvascular transport via the EPR effect, (3) interstitial diffusion and convection, (4) cellular binding and internalization, and (5) drug release kinetics. Model predictions are validated against experimental measurements of intratumoral drug concentration profiles obtained via microdialysis, mass spectrometry imaging, or fluorescent tracer studies [93].
Comparative Performance Analysis of Computational Platforms
Table 3: Quantitative Comparison of Computational Design Approaches
| Performance Metric | Molecular Dynamics | AI/Machine Learning | Multi-Scale Modeling |
|---|---|---|---|
| Spatial Resolution | Atomic-scale (Å) [95] | Nanoscale to system-level [95] | Cellular to organ-level [93] |
| Temporal Range | Nanoseconds to microseconds [95] | N/A (static predictions) | Minutes to days [93] |
| Throughput | Low to moderate (days to weeks/simulation) [95] | High (seconds once trained) [96] | Moderate (hours to days) [93] |
| Predictive Accuracy | High for molecular interactions [95] | Variable (depends on training data quality) [95] | Moderate to high for biodistribution [93] |
| Experimental Correlation | R² ~0.7-0.9 for membrane penetration [95] | R² ~0.8-0.95 for formulation properties [96] | R² ~0.6-0.8 for tumor accumulation [93] |
| Key Limitations | System size and timescale constraints [95] | Data scarcity and quality issues [95] | Simplified biology and parameter uncertainty [93] |
Integrated Workflows and Experimental Translation
The most impactful applications emerge from integrating multiple computational approaches into cohesive workflows. For instance, MD simulations can provide atomic-level insights for training AI models, while multi-scale simulations can contextualize AI predictions within physiological environments [95] [9].
Case Study – Optimized ThermoDox Delivery: Research demonstrates the power of computational integration for improving thermally-activated liposomal doxorubicin (ThermoDox) [93]. Multi-scale modeling revealed that conventional administration methods achieved only minimal tumor penetration due to elevated interstitial fluid pressure in solid tumors. Simulations predicted that an intravascular release paradigm, where thermosensitive nanoparticles release their payload within tumor vasculature upon mild heating (41-42°C), could significantly improve drug bioavailability. This approach increased predicted intracellular drug concentration by 3.7-fold compared to conventional extravasation-dependent delivery [93].
Table 4: Key Research Reagents and Computational Resources for Targeting Nanosystem Development
| Category | Specific Examples | Function in Research | Considerations for Selection |
|---|---|---|---|
| Simulation Software | GROMACS, AMBER, LAMMPS [95] | MD simulation execution and analysis | Force field compatibility; Scalability to large systems [95] |
| AI/ML Platforms | TensorFlow, PyTorch, Scikit-learn [96] | Developing predictive models for nanoparticle design | Integration with cheminformatics pipelines; Hyperparameter optimization [96] |
| Nanoparticle Libraries | Liposomes, PLGA, gold nanoparticles, dendrimers [4] [9] | Experimental validation of computational predictions | Biocompatibility; Drug loading capacity; Scalability [4] |
| Targeting Ligands | Antibodies, peptides, aptamers, folic acid [9] [91] | Functionalization for active targeting | Binding affinity; Immunogenicity; Conjugation efficiency [91] |
| Characterization Tools | DLS, NTA, TEM, HPLC [4] | Physicochemical property measurement | Resolution limits; Sample preparation requirements [4] |
Future Directions and Concluding Perspectives
The convergence of computational modeling and AI-assisted design is fundamentally transforming the development of targeting nanosystems for cancer therapy. These in silico approaches have demonstrated remarkable capabilities in optimizing nanoparticle physicochemical properties, predicting biological interactions, and ultimately improving therapeutic outcomes. The most promising path forward lies in the systematic integration of these complementary technologies into standardized workflows that leverage their respective strengths while acknowledging their limitations [95] [9].
As the field advances, key challenges remain in improving model biological fidelity, expanding experimental validation datasets, and enhancing computational accessibility for non-specialists. The successful translation of these computational tools into clinically viable nanotherapeutics will require continued collaboration between computational scientists, experimental researchers, and clinical oncologists. With rapid advancements in computing power, algorithm sophistication, and biological understanding, computational design is poised to become an indispensable component of the next generation of targeted cancer nanomedicines.
Quantitative Assessment and Efficacy Comparison of Targeting Approaches
In Vitro and In Vivo Models for Evaluating Nanoparticle Targeting Efficiency
The journey of a targeted nanoparticle from the laboratory bench to the clinic is fraught with biological complexities. A therapeutic formulation that demonstrates exceptional cancer cell specificity in a controlled laboratory environment may fail to accumulate in tumors in a living organism due to unforeseen immune interactions, physiological barriers, or off-target clearance [25]. This disconnect between experimental models represents one of the most significant hurdles in nanomedicine development. Selecting appropriate and predictive models for evaluating nanoparticle targeting efficiency is therefore not merely a technical choice but a fundamental determinant of translational success. This guide provides a comprehensive comparison of contemporary in vitro and in vivo models, detailing their applications, limitations, and integrated use within the broader context of optimizing nanoparticle targeting for cancer therapy.
Comparative Analysis of Evaluation Models
The following table summarizes the core characteristics, advantages, and limitations of the primary models used in the field. A sophisticated research program typically leverages data from multiple models to build a robust understanding of nanoparticle behavior.
Table 1: Comparison of In Vitro and In Vivo Models for Nanoparticle Targeting Evaluation
| Model Type | Key Characteristics | Measured Outcomes | Advantages | Disadvantages |
|---|---|---|---|---|
| 2D Monolayer Cultures [25] | - Single cell type grown on a flat surface- Simple, high-throughput- Controlled environment | - Cellular uptake (e.g., via ICP-MS)- Binding specificity- Cytotoxicity | - Low cost, high reproducibility- Rapid screening of formulations- Direct mechanism study | - Lacks tissue structure and complexity- Poor predictor of in vivo penetration and efficacy |
| 3D Spheroid Models [25] | - Multicellular aggregates mimicking micro-tumors- Develops nutrient/oxygen gradients | - Nanoparticle penetration depth- Uptake in different cell layers | - Models diffusion barriers and ECM- More physiologically relevant than 2D | - Does not recapitulate immune system or systemic clearance |
| Immunocompromised In Vivo Models (e.g., nude mice) | - Lack functional T-cells and sometimes B-cells- Support human tumor xenografts | - Biodistribution (e.g., organ accumulation)- Tumor-specific targeting- Therapeutic efficacy | - Allows study of human-derived tumors- Reduces graft-versus-host disease | - Fails to capture critical immune interactions (e.g., MPS clearance) [25] |
| Immunocompetent Syngeneic In Vivo Models [25] | - Intact immune system- Tumors derived from the same genetic background | - Biodistribution in a full immune context- Immune-driven off-target clearance (liver, spleen)- Realistic therapeutic index | - Provides physiologically relevant assessment of targeting- Accounts for immune clearance | - Does not involve human tumor biology- Can be more variable |
Quantitative data starkly highlights the model-dependent nature of results. For instance, a 2025 study found that RGD-functionalized gold nanoparticles exhibited an impressive ~150-fold increase in cellular uptake in monolayer cultures of KPCY murine pancreatic cancer cells compared to non-targeted particles [25]. However, this dramatic in vitro success did not translate in vivo. When tested in immunocompetent mice, the same RGD-targeted nanoparticles showed significantly reduced tumor accumulation due to enhanced off-target clearance by the mononuclear phagocyte system (MPS), with increased sequestration in the liver and spleen [25]. This critical finding would have been entirely missed in an immunocompromised model.
Experimental Protocols for Key Assays
Protocol: Evaluating Targeted Uptake in 3D Spheroids
This protocol assesses a nanoparticle's ability to penetrate beyond superficial cell layers, a crucial capability for solid tumor targeting [25].
- Spheroid Generation: Seed KPCY murine pancreatic cancer cells (or other relevant line) in agarose-coated 96-well plates to promote unconstrained 3D aggregation. Culture until spheroids reach a diameter of 300-500 µm.
- Nanoparticle Exposure: Incubate mature spheroids with targeted nanoparticles (e.g., GNP-PEG-cRGD) and non-targeted controls (e.g., GNP-PEG) at a concentration of 7.5 µg/mL for 1, 8, and 24 hours.
- Sample Processing and Analysis:
- Option A (Quantitative): Harvest spheroids at each time point (n=5 per group). Digest tissues using aqua regia and quantify intracellular gold content via Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
- Option B (Qualitative): Fix spheroids, embed in OCT compound, and section. Image using confocal microscopy (if nanoparticles are fluorescently tagged) to visualize and compare penetration depth and intracellular accumulation between targeted and non-targeted formulations.
Protocol: Biodistribution in Immunocompetent Models
This protocol provides a physiologically relevant assessment of tumor targeting efficiency and systemic clearance [25].
- Animal and Tumor Model: Establish syngeneic tumors in immunocompetent C57BL/6 mice by subcutaneously injecting KPCY cells. Proceed with studies when tumors reach a volume of ~100 mm³.
- Dosing and Sample Collection:
- Administer a single intravenous dose (e.g., 5 mg/kg gold) of the targeted nanoparticle formulation and a non-targeted control to separate groups of tumor-bearing mice (n=6-8 per group).
- At predetermined time points (e.g., 1, 4, 24, 48 hours) post-injection, euthanize animals and collect major organs (tumor, liver, spleen, kidneys, heart, lungs) and blood.
- Quantification of Accumulation: Digest weighed tissue samples in concentrated nitric acid and hydrogen peroxide at 70°C. Dilute the digested samples and analyze via ICP-MS to determine the gold concentration, normalized to tissue weight (% injected dose per gram, %ID/g). This allows direct comparison of tumor targeting versus off-target accumulation.
Integrated Workflow and Emerging Technologies
A rational workflow for evaluating nanoparticle targeting begins with high-throughput in vitro screens but must prioritize immunocompetent in vivo models late in the development pipeline to de-risk subsequent translational steps [25]. Advanced technologies are now augmenting traditional models.
Artificial intelligence (AI) and machine learning (ML) are emerging as transformative tools. By analyzing large, high-dimensional datasets, AI-driven models can predict nanoparticle biodistribution, model complex nano-bio interactions like protein corona formation, and guide optimal nanocarrier design, thereby reducing reliance on resource-intensive trial-and-error experiments [97]. Furthermore, single-particle characterization technologies (e.g., mass photometry) are addressing critical quality control challenges. These methods move beyond traditional ensemble-average measurements to quantify therapeutic loading and targeting ligand coating efficiencies at the single-nanoparticle level, providing crucial insights into batch heterogeneity that can profoundly affect therapeutic outcomes [98].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Materials for Nanoparticle Targeting Studies
| Reagent/Material | Function in Evaluation | Specific Examples |
|---|---|---|
| Syngeneic Cell Lines [25] | Forms tumors in immunocompetent hosts with intact immune context for relevant in vivo testing. | KPCY murine pancreatic cancer cells. |
| Targeting Ligands | Confers specificity to nanoparticles by binding overexpressed receptors on target cancer cells. | RGD peptides (linear & cyclic) for ανβ3 integrin [25]; Anti-Fc nanobodies for antibody-oriented targeting [99]. |
| PEGylated Lipids | Imparts "stealth" properties by reducing protein adsorption, extending circulation half-life, and improving passive targeting via the EPR effect. | DMG-PEG2000, DSPE-PEG2000 [99]. |
| Ionizable Lipids | Key structural and functional component of lipid nanoparticles (LNPs), critical for encapsulating nucleic acid payloads (e.g., mRNA). | DLin-MC3-DMA (MC3), SM-102 [99]. |
| Characterization Tools | Measures hydrodynamic size, concentration, and surface charge of nanoparticles to ensure batch consistency. | Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA) [98]. |
| Analytical Instruments | Provides quantitative, gold-standard data on nanoparticle material (e.g., gold, silica) uptake in cells and tissues. | Inductively Coupled Plasma Mass Spectrometry (ICP-MS) [25]. |
In the pursuit of a "magic bullet" for cancer therapy, nanoparticle-based drug delivery systems have emerged as a transformative approach, primarily utilizing two distinct strategies: passive and active targeting [100] [101]. Passive targeting leverages the inherent physiological and pathological characteristics of tumors, while active targeting employs specific ligand-receptor interactions to enhance tumor selectivity [102]. For researchers and drug development professionals, understanding the efficacy, limitations, and appropriate application contexts of these strategies is crucial for designing effective nanotherapeutics. This guide provides a direct comparison of these approaches within preclinical models, synthesizing current experimental data, methodologies, and key considerations to inform research and development decisions. The fundamental distinction lies in their mechanisms: passive targeting depends on the enhanced permeability and retention (EPR) effect for tumor accumulation, whereas active targeting facilitates specific cellular binding and internalization through surface-functionalized ligands [100] [101] [103].
Core Mechanisms and Fundamental Principles
Passive Targeting: Exploiting the EPR Effect
The foundational principle of passive targeting is the Enhanced Permeability and Retention (EPR) effect, first described by Maeda and Matsumura [100]. This phenomenon arises from two key pathological features of solid tumors:
- Leaky Vasculature: Tumor blood vessels are highly disorganized, dilated, and contain wide endothelial gaps (often ≥600 nm) due to rapid, defective angiogenesis. This allows substantial extravasation of nanoparticles from the bloodstream into the tumor interstitium [100].
- Impaired Lymphatic Drainage: Tumors frequently lack a well-developed lymphatic system, leading to reduced clearance and prolonged retention of accumulated nanoparticles [100].
The EPR effect is significantly influenced by nanoparticle characteristics. Size is particularly critical; an optimal diameter of 20 to 200 nm favors prolonged circulation, enhances tumor accumulation, and reduces renal elimination (which affects particles <7 nm) and hepatic clearance [102]. Surface properties also play a vital role; PEGylation, the conjugation of polyethylene glycol, creates a hydrophilic layer that minimizes opsonization and recognition by the mononuclear phagocyte system (MPS), thereby extending circulation half-life and increasing the likelihood of tumor deposition [104] [102].
Active Targeting: Leveraging Molecular Recognition
Active targeting enhances the specificity of nanoparticles by decorating their surface with biorecognition molecules (ligands) that bind to receptors overexpressed on target cancer cells or within the tumor vasculature [101] [103]. This strategy aims to overcome the primary limitation of passive targeting—the inability to differentiate between different cell types once within the tumor interstitium.
The process typically involves:
- Ligand-Receptor Binding: Specific interaction between the nanoparticle's surface ligands (e.g., antibodies, peptides, aptamers, small molecules) and corresponding cell surface receptors [101] [103].
- Receptor-Mediated Endocytosis: The binding event triggers cellular internalization of the nanoparticle, facilitating efficient intracellular drug delivery [101].
This approach not only improves cellular uptake but can also suppress multidrug resistance (MDR) mechanisms, such as P-glycoprotein-mediated drug efflux, by bypassing certain resistance pathways [101].
Table 1: Common Targeting Ligands and Their Corresponding Receptors
| Ligand Type | Target Receptor | Cancer Type(s) with Receptor Overexpression | Key Findings (Preclinical) |
|---|---|---|---|
| Trastuzumab (mAb) | HER2 [101] | Breast (∼25% of invasive cases) [101] | Specific targeting of HER2+ cells; used for imaging and drug delivery [101]. |
| Cetuximab (mAb) | EGFR [101] [103] | Pancreatic, Colorectal, Lung [101] [103] | Targeted gold nanoparticles enabled thermal ablation of pancreatic xenografts [101]. |
| RGD Peptide | αvβ3 Integrin [25] [103] | Glioblastoma, Lung, Esophageal [103] | Improves cellular uptake in vitro; in vivo efficacy is highly model-dependent [25] [103]. |
| Folic Acid | Folate Receptor [101] | Ovarian, Lung, Breast [101] | Not detailed in search results, but widely used in targeted nanomedicine. |
| Anti-VCAM Antibody | VCAM-1 [105] | Inflamed Endothelium (e.g., in Stroke Models) [105] | LNPs targeted to brain vasculature showed ~4x higher brain distribution [105]. |
The following diagram illustrates the sequential relationship between passive and active targeting in a typical nanoparticle journey to a tumor cell.
Diagram 1: The sequential journey of a targeted nanoparticle, highlighting the distinct phases of passive accumulation via the EPR effect and subsequent active, ligand-receptor mediated cellular binding and internalization.
Comparative Efficacy Analysis in Preclinical Models
Quantitative Comparison of Tumor Accumulation
Evaluating the success of nanoparticle delivery involves measuring how much of the administered dose reaches the tumor. The table below summarizes key quantitative findings from preclinical studies comparing passive and active targeting strategies.
Table 2: Preclinical Efficacy Metrics: Passive vs. Active Targeting
| Nanoparticle Type / Strategy | Tumor Model | Key Efficacy Metric | Reported Outcome | Citation |
|---|---|---|---|---|
| PEGylated Gold NPs (Passive) | KPCY (Pancreatic) | Tumor Accumulation | Baseline for comparison | [25] |
| RGD-functionalized Gold NPs (Active) | KPCY (Pancreatic) | Tumor Accumulation | ▼ Significant reduction vs. passive (due to MPS clearance) | [25] |
| VCAM-targeted LNPs (Active) | Experimental ICH (Stroke) | Brain Distribution | ▲ ~4x higher than non-specific LNPs | [105] |
| RGD-HSA NPs (Resveratrol) | Not Specified | Cellular Internalization | ▲ ~3.6-fold higher in vitro | [103] |
| RGD-NPs (PTX & Cisplatin) | Lung Cancer | Cellular Endocytosis | ▲ ~1.4-fold higher in vitro | [103] |
Impact of the Immunocompetent Tumor Microenvironment
A critical factor emerging from recent studies is the profound impact of the host immune system on targeting efficacy, which is often overlooked in traditional immunocompromised models.
- Immunocompromised vs. Immunocompetent Models: Many studies demonstrating the success of active targeting ligands like RGD peptides have been conducted in immunodeficient mouse models (e.g., xenografts) [25]. These models lack a fully functional immune system, potentially leading to an overestimation of targeting efficacy because they do not account for immune-mediated clearance of nanoparticles [25] [100].
- The MPS Hurdle: A 2025 study using an immunocompetent syngeneic tumor model (KPCY) revealed that while RGD functionalization significantly increased GNP uptake in cancer cells in vitro, it significantly reduced tumor accumulation in vivo [25]. This counterintuitive result was attributed to enhanced off-target clearance by the mononuclear phagocyte system (MPS), with elevated accumulation observed in the spleen and liver [25]. This highlights that a ligand's ability to improve cellular uptake does not guarantee successful tumor delivery in vivo, as it may also increase recognition by immune cells.
Experimental Protocols for Direct Comparison
To ensure a valid direct comparison between passive and active targeting strategies in preclinical studies, consistent and well-controlled experimental designs are essential.
Protocol for Synthesis and Functionalization of Targeted Nanoparticles
Example: Gold Nanoparticle (GNP) Functionalization with RGD Peptides [25]
- Synthesis of Core NPs: Synthesize spherical gold nanoparticles (~12 nm core diameter) using the citrate reduction method. Characterize size and morphology via Transmission Electron Microscopy (TEM).
- PEGylation (Stealth Layer): Functionalize citrate-capped GNPs with a dense layer of thiol-terminated PEG (e.g., 2 kDa) at a specific grafting density (e.g., 1 PEG molecule per nm² of GNP surface). This step is crucial for enhancing colloidal stability and prolonging circulation time for both passive and active strategies.
- Ligand Conjugation (Active Targeting): For the active targeting group, conjugate RGD peptides (linear or cyclic) to the PEGylated GNPs. Use a cysteine-terminated RGD peptide for thiol-based chemistry. A common ratio is 1 RGD peptide per 2 PEG molecules.
- Characterization: After each functionalization step, characterize the nanoparticles using:
- Dynamic Light Scattering (DLS): To measure hydrodynamic diameter and polydispersity index (PDI).
- Zeta Potential: To verify surface charge changes indicative of successful conjugation.
- UV-Vis Spectroscopy: To monitor shifts in surface plasmon resonance peak.
Protocol for In Vivo Biodistribution and Efficacy Studies
- Animal and Tumor Model Selection:
- Use immunocompetent syngeneic models (e.g., KPCY for pancreatic cancer) where possible to most accurately recapitulate the human immune-tumor interactions [25].
- Compare with traditional xenograft models in immunodeficient mice to contextualize findings.
- Dosing and Administration: Administer nanoparticles (e.g., 7.5 µg GNP/g body weight) intravenously to tumor-bearing mice. Ensure the encapsulated drug dose is equivalent across all experimental groups.
- Biodistribution Analysis:
- At predetermined time points (e.g., 1, 8, 24 hours post-injection), euthanize animals and collect tumors and major organs (liver, spleen, kidneys, heart, lungs).
- Use Inductively Coupled Plasma Mass Spectrometry (ICP-MS) to quantitatively determine the gold content (for metal NPs) or a radioactive/fluorescent label in each tissue.
- Calculate the % Injected Dose per Gram of tissue (%ID/g) to compare accumulation.
- Efficacy Assessment:
- Monitor tumor volume over time.
- At study endpoint, analyze survival rates and collect tumors for histological analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for Nanoparticle Targeting Studies
| Reagent / Material | Function / Role | Example from Literature |
|---|---|---|
| Ionizable Lipids (e.g., MC3) | Forms core of LNPs; enables nucleic acid encapsulation and endosomal escape [102]. | Key component in FDA-approved Onpattro [102]. |
| DSPE-PEG Lipids | Provides "stealth" properties; reduces protein adsorption and MPS clearance; can be functionalized for ligand attachment [102] [105]. | Used in Doxil and modern mRNA LNPs [102] [105]. |
| Targeting Ligands (e.g., cRGD, Cetuximab) | Mediates specific binding to overexpressed receptors on target cells (active targeting) [101] [25]. | cRGD for αvβ3 integrin; Cetuximab for EGFR [101] [25]. |
| Microfluidic Mixers (e.g., NanoAssemblr) | Enables reproducible, scalable synthesis of LNPs with controlled size and low PDI [102] [105]. | Used for formulating VCAM-targeted IL-10 mRNA LNPs [105]. |
| Dynamic Light Scattering (DLS) | Instrumentation to measure nanoparticle hydrodynamic size, distribution, and stability [25] [105]. | Standard for characterizing GNPs and LNPs pre- and post-functionalization [25] [105]. |
| Inductively Coupled Plasma Mass Spectrometry (ICP-MS) | Highly sensitive quantitative elemental analysis for biodistribution studies of metallic nanoparticles [25]. | Used to measure gold content in tissues from GNP-treated mice [25]. |
Integrated Discussion and Research Perspectives
The direct comparison between passive and active targeting reveals a nuanced landscape. Passive targeting, driven by the EPR effect, provides the foundational mechanism for initial tumor accumulation and is responsible for the clinical success of nanomedicines like Doxil and Abraxane [104]. However, its efficacy is highly variable across tumor types and is influenced by factors such as tumor vasculature and interstitial pressure [104] [106].
Active targeting excels at enhancing cellular internalization once nanoparticles have extravasated into the tumor interstitium, as consistently demonstrated in vitro [101] [103]. The critical caveat is that this benefit does not always translate to increased overall tumor accumulation in vivo, particularly in immunocompetent models [25]. The ligand itself can alter the protein corona and nanoparticle pharmacokinetics, potentially accelerating clearance by the MPS and thereby reducing the number of nanoparticles available to reach the tumor [25]. This underscores that active targeting should be viewed as a complement to, not a replacement for, effective passive targeting. The optimal strategy is a sequential process: nanoparticles must first have long circulation times to leverage the EPR effect (passive targeting) before the ligands can mediate specific binding to cancer cells (active targeting).
Future research directions should focus on:
- Patient Stratification: Identifying biomarkers predictive of a strong EPR effect in patients to enrich clinical trial populations likely to respond to nanotherapeutics [104].
- Advanced Ligand Engineering: Developing ligands with minimal immunogenicity and optimized binding properties to balance target affinity with MPS evasion.
- Model Fidelity: Prioritizing the use of immunocompetent models and incorporating tumor microenvironment modulators (e.g., angiotensin II to enhance EPR) in preclinical studies to improve clinical translatability [100].
In conclusion, the choice between passive and active targeting is not a binary one. The most promising path forward lies in the intelligent integration of both strategies, where nanoparticles are engineered for prolonged circulation and efficient EPR-mediated tumor accumulation, and then equipped with ligands to facilitate precise cellular uptake and drug delivery within the tumor.
Quantitative Analysis of Tumor Accumulation and Cellular Uptake Rates
The success of nanoparticle-based cancer therapeutics hinges on their efficient delivery to tumor sites, a process governed by two critical pharmacokinetic phases: (1) tumor accumulation, the passive or active transport of nanoparticles from systemic circulation into the tumor interstitium, and (2) cellular uptake, the subsequent internalization of nanoparticles by cancer cells following their extravasation [25] [1]. Rapid advancements in nanotechnology have yielded a diverse array of nanocarriers, including lipid-based, polymeric, and inorganic nanoparticles, designed to improve drug solubility, prolong circulation, and control drug release [7] [1]. However, the clinical translation of these sophisticated nanomedicines remains limited, with only a few formulations successfully advancing from preclinical studies to clinical approval [1]. A significant translational barrier lies in the complex and often unpredictable interplay between nanoparticle design and the dynamic tumor microenvironment (TME), which can substantially alter expected targeting outcomes [107]. This guide provides a quantitative, data-driven comparison of nanoparticle performance, focusing on how specific physicochemical properties and targeting strategies influence the fundamental metrics of tumor accumulation and cellular uptake.
Comparative Performance Data of Nanoparticle Targeting
The efficacy of nanoparticle targeting is quantified through key metrics such as tumor accumulation efficiency, often measured as the percentage of injected dose per gram of tissue (%ID/g), and cellular uptake rates, determined via techniques like inductively coupled plasma mass spectrometry (ICP-MS) for inorganic nanoparticles or fluorescence-activated cell sorting (FACS) for fluorescently tagged carriers [25]. The data reveal that performance is not absolute but is highly dependent on nanoparticle design and tumor stage.
Table 1: Comparative Tumor Accumulation of Actively Targeted vs. Passively Targeted Gold Nanoparticles (AuNPs)
| Nanoparticle Type | Size (nm) | Targeting Ligand | Tumor Model | Key Finding on Tumor Accumulation | Key Finding on Cellular Uptake (In Vitro) |
|---|---|---|---|---|---|
| Active AuNPs [107] | 7, 15, 45, 90 | RGD (cyclic) | Orthotopic Breast Cancer (different stages) | Size- and stage-dependent: 7 nm NPs best in early tumors; 90 nm NPs best in late-stage tumors with high integrin αvβ3. | N/A |
| Passive AuNPs [107] | 7, 15, 45, 90 | None (PEG-only) | Orthotopic Breast Cancer | Lower accumulation compared to active AuNPs across all sizes and tumor stages. | N/A |
| Active GNPs [25] | ~12 (core) | RGD (linear & cyclic) | Immunocompetent Syngeneic (KPCY) | Reduced accumulation vs. PEG-only due to enhanced off-target clearance by the Mononuclear Phagocyte System (MPS). | Significantly increased: ~100-150 fold increase at 1h with RGD. |
| Passive GNPs [25] | ~12 (core) | None (PEG-only) | Immunocompetent Syngeneic (KPCY) | Higher accumulation than RGD-functionalized GNPs. | Served as baseline for uptake measurements. |
Table 2: Impact of Tumor Stage and Pathophysiology on Nanoparticle Accumulation Data derived from a study on RGD-modified AuNPs in an orthotopic breast cancer model [107].
| Tumor Stage | Pathophysiological Characteristics | Optimal NP Size | Dominant Accumulation Mechanism |
|---|---|---|---|
| Early Stage | Looser ECM, larger gaps, lower interstitial fluid pressure, less receptor expression. | 7 nm | Size-dependent penetration dominates. |
| Mid Stage | Transitional pathophysiology. | 7 nm and 90 nm | Balanced influence of size effects and multivalent interactions. |
| Late Stage | Denser ECM, higher interstitial fluid pressure, highly expressed target receptors (integrin αvβ3). | 90 nm | Multivalent binding between larger NPs and receptors dominates. |
Experimental Protocols for Assessing Targeting Efficiency
Robust and standardized experimental methodologies are imperative for generating reliable, comparable quantitative data on nanoparticle targeting. The following sections detail key protocols cited in the comparative data.
Synthesis and Functionalization of Gold Nanoparticles (AuNPs)
Application: This protocol is used to create the core nanoparticles, such as the RGD-modified and PEGylated AuNPs featured in the comparison tables [25] [107].
Detailed Methodology:
- Synthesis of Citrate-Capped AuNPs: Spherical gold nanoparticles with an average core diameter of approximately 12-15 nm are synthesized via the citrate reduction method. An aqueous solution of hydrogen tetrachloroaurate (HAuCl₄) is brought to a boil under vigorous stirring, followed by the rapid addition of a sodium citrate solution. The solution color changes from yellow to deep red, indicating nanoparticle formation [25] [107].
- PEGylation for Stealth Properties: Citrate-capped AuNPs are functionalized with thiol-terminated methoxy-polyethylene glycol (mPEG-SH, typically 2-3.5 kDa) at a specific grafting density (e.g., 1 PEG molecule per nm² of GNP surface area) to form GNP-PEG. This step enhances colloidal stability, reduces nonspecific protein adsorption, and extends blood circulation time [25] [107].
- Conjugation of Targeting Ligands: For active targeting, RGD peptides (linear or cyclic) containing a cysteine residue at the N-terminus are conjugated to the PEG-coated nanoparticles. This is achieved either by co-incubation with mPEG-SH or by using heterobifunctional PEG (e.g., NHS-PEG-SH). The conjugation is confirmed through characterization techniques [25] [107].
Critical Characterization Steps:
- Size and Morphology: Transmission Electron Microscopy (TEM) is used to determine the core size and shape.
- Hydrodynamic Size and Stability: Dynamic Light Scattering (DLS) measures the hydrodynamic diameter in solution, which increases with successive PEG and RGD conjugation.
- Surface Charge: Zeta potential analysis tracks the change in surface charge from negative (citrate-capped) to neutral (PEGylated) and a subsequent shift upon RGD attachment.
- Optical Properties: UV-Vis spectroscopy confirms nanoparticle stability and monitors functionalization through shifts in the surface plasmon resonance peak [25] [108].
In Vitro Cellular Uptake Assay
Application: This protocol is used to quantify the internalization of nanoparticles by cancer cells, as demonstrated by the ~100-150 fold increase in uptake of RGD-functionalized GNPs [25].
Detailed Methodology:
- Cell Culture: Cancer cells (e.g., KPCY murine pancreatic cancer cells) are cultured as either 2D monolayers or 3D spheroids to better mimic the tumor microenvironment.
- Nanoparticle Exposure: Cells are exposed to various GNP complexes (e.g., GNP-PEG, GNP-PEG-lRGD, GNP-PEG-cRGD) at a predetermined concentration (e.g., 7.5 µg/mL) for set time intervals (e.g., 1 h, 8 h, 24 h).
- Sample Harvesting and Processing: At each time point, cells are washed thoroughly with buffer to remove non-internalized nanoparticles, trypsinized, and counted.
- Quantification of Uptake: The intracellular gold content is quantified using Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Data are typically normalized to the number of cells or total protein content. Uptake can be further verified qualitatively using confocal microscopy for fluorescently labeled nanoparticles [25].
In Vivo Biodistribution and Tumor Accumulation Study
Application: This protocol is used to evaluate the real-world targeting efficiency of nanoparticles in a living organism, revealing critical insights such as the immune-driven clearance of RGD-functionalized particles [25].
Detailed Methodology:
- Animal and Tumor Model: Immunocompetent mice bearing syngeneic tumors (e.g., the KPCY model) are used to provide a physiologically relevant assessment of immune interactions, unlike immunocompromised models which can overestimate efficacy [25].
- Dosing and Administration: Nanoparticles are administered intravenously via the tail vein at a specific dose.
- Tissue Collection and Analysis: After a predetermined circulation time (e.g., 24 h or 48 h), animals are euthanized, and tissues of interest (tumor, liver, spleen, kidneys, etc.) are collected, weighed, and digested.
- Quantification of Biodistribution: The gold content in each digested tissue sample is measured using ICP-MS. Tumor accumulation is calculated as the percentage of the injected dose per gram of tissue (%ID/g). Comparative analysis of organ distribution reveals off-target accumulation, particularly in the liver and spleen, which are organs of the Mononuclear Phagocyte System (MPS) [25].
Visualizing Signaling Pathways and Experimental Workflows
The following diagrams illustrate the critical signaling pathways involved in active targeting and the experimental workflow for evaluating nanoparticle efficacy.
Diagram 1: RGD-Integrin Targeting Pathway and Outcomes. This diagram contrasts the in vitro pathway, where RGD binding to integrin αvβ3 enhances cellular uptake, with the in vivo outcome, where the same interaction can promote immune recognition and reduce tumor accumulation [25] [107] [109].
Diagram 2: Experimental Workflow for NP Evaluation. This workflow outlines the standard pipeline for synthesizing, characterizing, and testing nanoparticle targeting efficiency from in vitro to in vivo settings [25] [107] [108].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table catalogues key reagents and materials essential for conducting experiments in nanoparticle tumor targeting, as derived from the methodologies cited in this guide.
Table 3: Essential Research Reagents and Materials for Nanoparticle Targeting Studies
| Reagent/Material | Function and Application in Research | Example from Literature |
|---|---|---|
| Gold Salts (e.g., HAuCl₄) | Precursor for the synthesis of gold nanoparticle (AuNP) cores. | Used in the synthesis of 7-90 nm AuNPs for size-dependent accumulation studies [107]. |
| Functional PEGs (e.g., mPEG-SH, NHS-PEG-SH) | Imparts "stealth" properties to reduce immune clearance; serves as a linker for conjugating targeting ligands. | Conjugated to AuNPs to form a passive targeting "PEG-only" control and as a backbone for RGD attachment [25] [107]. |
| RGD Peptides (linear & cyclic) | Targeting ligand that binds to the ανβ3 integrin receptor overexpressed on tumor cells and vasculature. | Functionalized onto PEGylated GNPs/AuNPs to create active targeting nanoparticles [25] [107]. |
| Cell Lines (e.g., KPCY) | In vitro models for evaluating cellular uptake mechanisms and efficacy. | KPCY murine pancreatic cancer cells used in monolayer and 3D spheroid uptake assays [25]. |
| Syngeneic Tumor Models | Immunocompetent animal models for physiologically relevant in vivo biodistribution studies. | KPCY model used to demonstrate RGD-mediated MPS clearance, a finding absent in immunodeficient models [25]. |
| ICP-MS (Inductively Coupled Plasma Mass Spectrometry) | Highly sensitive analytical technique for the quantitative measurement of elemental metals (e.g., gold) in cells and tissues. | Used to quantify intracellular GNP uptake in vitro and GNP accumulation in tumors and organs in vivo [25]. |
| TEM (Transmission Electron Microscopy) | High-resolution imaging technique for determining the core size, morphology, and distribution of nanoparticles. | Used to confirm the ~12 nm core diameter of synthesized citrate-capped GNPs [25] [108]. |
The efficacy of nanoparticle-based cancer therapies is fundamentally governed by their targeting efficiency—the ability to accumulate therapeutic cargo specifically at the tumor site while minimizing off-target distribution. This parameter directly influences therapeutic outcomes, particularly tumor growth inhibition, by determining the effective drug dose delivered to cancer cells [35] [110]. The pursuit of enhanced targeting has driven innovation in nanocarrier design, leading to diverse strategies including passive targeting via the Enhanced Permeability and Retention (EPR) effect, active targeting using surface-bound ligands, and stimulus-responsive drug release mechanisms [35] [34]. However, the correlation between improved cellular uptake observed in vitro and subsequent tumor growth inhibition in vivo is not always straightforward, being influenced by complex biological barriers such as the tumor microenvironment and the mononuclear phagocyte system (MPS) [25]. This guide provides a comparative analysis of leading nanoparticle targeting strategies, evaluating their performance through the critical lens of experimental tumor growth inhibition data, and offers detailed methodologies for assessing their efficacy in preclinical models.
Comparative Analysis of Nanoparticle Targeting Strategies
The landscape of tumor-targeted nanoparticles is diverse, encompassing various carrier types and targeting moieties, each with distinct strengths and limitations. The following analysis compares key strategies based on their design, targeting mechanism, and demonstrated efficacy in tumor growth inhibition.
Table 1: Comparison of Nanoparticle Targeting Strategies and Therapeutic Outcomes
| Nanoparticle Type & Targeting Moisty | Target Receptor/Mechanism | Key Experimental Findings | Reported Tumor Growth Inhibition | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| cRGD-functionalized Gold Nanoparticles (Active) [25] | ανβ3 Integrin | Reduced in vivo tumor accumulation vs. non-targeted PEG-NPs in immunocompetent models; enhanced MPS clearance. | Not directly measured; reduced accumulation suggests potential compromise. | High specificity in in vitro models. | Efficacy is model-dependent; can enhance immune clearance. |
| CPMycNB Nanobody (Active) [111] | c-MYC Oncoprotein | Disrupted c-MYC/MAX interaction in nucleus; downregulated target genes; induced apoptosis. | Significant reduction in tumor size and weight in xenograft models. | Targets "undruggable" intracellular oncoprotein; direct mechanism. | Requires cell and nuclear penetration; delivery challenges. |
| Peptide-Targeted Ultrasound Nanoparticle [112] | Tumor-Homing Peptide + Focused Ultrasound | Combined mechanical ablation with chemotherapeutic "one-two punch"; 100-fold reduced ultrasound energy requirement. | Complete tumor disappearance in some melanoma models; improved 60-day survival. | Synergistic physical & chemical therapy; spatiotemporal control. | Platform complexity; requires specialized equipment. |
| HSA Nanoparticles (Passive/Active) [35] | gp60, SPARC (Passive: EPR effect) | Tumor enrichment up to 624 hours; validated in FDA-approved Abraxane. | Clinically validated efficacy in multiple cancers. | Excellent biocompatibility; proven clinical translation. | Limited human serum availability; moderate intrinsic specificity. |
| Chitosan Nanoparticles (Passive) [35] | EPR effect | Dose-dependent cardiotoxicity and embryotoxicity; high non-specific accumulation. | Limited by low targeting efficiency and off-target toxicity. | Biodegradable; mucoadhesive. | Low tumor targeting; significant off-target toxicity. |
The data indicates that while active targeting strategies like the c-MYC nanobody show high promise through direct engagement of oncogenic drivers, their success is contingent on efficient delivery to the intracellular target site [111]. Conversely, even well-designed active strategies like RGD-targeting can falter in vivo due to unforeseen biological interactions, such as accelerated clearance by the immune system [25]. Hybrid approaches that combine targeting with external stimuli, such as ultrasound, demonstrate how synergistic effects can lead to potent tumor growth inhibition, sometimes achieving complete regression [112].
Experimental Protocols for Evaluating Targeting and Efficacy
To reliably correlate targeting efficiency with tumor growth inhibition, standardized and rigorous experimental protocols are essential. Below are detailed methodologies for key assays used in the cited studies.
Protocol: In Vivo Biodistribution and Tumor Accumulation Study
This protocol, adapted from the evaluation of RGD-gold nanoparticles, quantifies how effectively nanoparticles localize to tumors versus other organs [25].
- Objective: To measure the pharmacokinetics and biodistribution of nanoparticles in a live tumor model.
- Materials:
- Immunocompetent mice (e.g., C57BL/6) with syngeneic tumors (e.g., KPCY pancreatic cancer model).
- Test nanoparticles (e.g., PEG-GNPs, RGD-PEG-GNPs).
- Anesthesia system (e.g., isoflurane).
- ICP-MS (Inductively Coupled Plasma Mass Spectrometry) system.
- Procedure:
- Administration: Inject nanoparticles intravenously into tumor-bearing mice via the tail vein at a standardized dose (e.g., equivalent Au dose per body weight).
- Time-Point Sampling: At predetermined time points (e.g., 1, 4, 8, 24, 48 hours), euthanize groups of mice and collect blood, tumor, and major organs (liver, spleen, kidneys, heart, lungs).
- Tissue Digestion: Digest the weighed tissue samples in concentrated nitric acid at elevated temperatures (e.g., 70°C overnight) to dissolve organic material and release nanoparticle cores.
- Analysis by ICP-MS: Dilute the digested samples and analyze them via ICP-MS to quantify the concentration of the nanoparticle core material (e.g., gold) in each tissue.
- Data Calculation: Calculate the percentage of injected dose per gram of tissue (%ID/g) for each sample. Plot the mean values over time to determine circulation half-life and compare final accumulation in the tumor versus off-target organs.
Protocol: In Vitro Cytotoxicity and Mechanism of Action Assay
This protocol is used to establish direct anti-proliferative effects and apoptotic induction, as demonstrated in the CPMycNB nanobody study [111].
- Objective: To assess the inhibitory effect of a therapeutic nanoparticle on cancer cell growth and proliferation and to elucidate its mechanism of action.
- Materials:
- Target cancer cell line (e.g., c-MYC-driven tumor cells).
- Cell culture media and reagents.
- MTT or CCK-8 cell viability assay kit.
- Annexin V-FITC / Propidium Iodide (PI) apoptosis detection kit.
- Flow cytometer.
- qRT-PCR equipment.
- Procedure:
- Cell Seeding: Seed cells into 96-well plates and allow to adhere overnight.
- Treatment: Treat cells with a concentration gradient of the therapeutic nanoparticle (e.g., CPMycNB) for 24-72 hours. Include negative and positive controls.
- Viability Measurement: Add MTT reagent and incubate. Measure the absorbance of the formed formazan crystals to determine cell viability and calculate IC50 values.
- Apoptosis Assay: Harvest treated cells, stain with Annexin V and PI, and analyze by flow cytometry to distinguish live, early apoptotic, late apoptotic, and necrotic cell populations.
- Gene Expression Analysis: Extract total RNA from treated cells. Perform qRT-PCR for key downstream target genes of the therapeutic target (e.g., c-MYC target genes) to confirm mechanism-specific activity.
Protocol: In Vivo Tumor Growth Inhibition Study
This is the cornerstone experiment for evaluating final therapeutic efficacy, as performed in the ultrasound nanoparticle and CPMycNB studies [111] [112].
- Objective: To evaluate the ability of a nanoparticle formulation to inhibit or regress tumor growth in a live animal model.
- Materials:
- Immunodeficient or immunocompetent mice with established subcutaneous xenografts or orthotopic tumors.
- Calipers for tumor measurement.
- Test nanoparticles, control nanoparticles, and free drug control.
- Procedure:
- Tumor Implantation: Implant cancer cells subcutaneously into the flank of mice.
- Randomization & Dosing: Once tumors reach a palpable size (e.g., ~100 mm³), randomize mice into treatment groups (e.g., saline control, free drug, non-targeted NP, targeted NP). Administer treatments via IV injection at set intervals (e.g., twice weekly for 3 weeks).
- Monitoring: Measure tumor dimensions (length and width) with calipers 2-3 times per week. Calculate tumor volume using the formula: Volume = (Length × Width²) / 2.
- Endpoint Analysis: Monitor animal body weight to assess toxicity. At the end of the study, harvest tumors, weigh them, and photograph them. Tumor growth inhibition is calculated as (1 - (Mean Tumor VolumeTreatment / Mean Tumor VolumeControl)) × 100%. Statistical significance is determined using tests like ANOVA.
Visualizing Key Biological Pathways and Workflows
Nanoparticle Tumor Targeting and Therapeutic Action Pathway
This diagram illustrates the multi-step journey of targeted nanoparticles from intravenous administration to final therapeutic outcome, integrating the key challenges and successes identified in the comparative analysis.
Experimental Workflow for Correlation Analysis
This workflow outlines the integrated experimental process, from nanoparticle characterization to final correlation analysis, providing a roadmap for researchers to generate comparable data.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful investigation into targeting efficiency and therapeutic outcomes relies on a suite of specialized reagents, biological models, and analytical instruments.
Table 2: Essential Research Reagents and Materials for Nanoparticle Evaluation
| Category | Item | Specific Example / Properties | Critical Function in Research |
|---|---|---|---|
| Nanoparticle Components | Polymeric Nanocarriers | PLGA, PEG, Chitosan [35] | Forms biodegradable, biocompatible nanoparticle core for drug encapsulation. |
| Targeting Ligands | RGD peptides [25], Nanobodies [111], Transferrin | Confers active targeting specificity to overexpressed tumor cell receptors. | |
| Stealth Agents | Polyethylene Glycol (PEG) of varying molecular weights [25] | Reduces opsonization and MPS clearance, extending circulation half-life. | |
| Biological Models | Cell Lines | KPCY murine pancreatic cells [25], c-MYC-driven tumor cells [111] | In vitro models for assessing targeting, uptake, and cytotoxicity. |
| 3D Spheroid Cultures | Multicellular tumor spheroids (MCTS) [25] | Provides a more physiologically relevant model for penetration studies than 2D monolayers. | |
| Animal Models | Immunocompetent syngeneic mice [25], Xenograft models [111] | Essential for in vivo biodistribution and efficacy studies in a whole-organism context. | |
| Analytical Instruments | ICP-MS | Inductively Coupled Plasma Mass Spectrometry [25] | Gold-standard for quantitative elemental analysis of metal-based NP biodistribution. |
| Dynamic Light Scattering | DLS / Zetasizer | Measures hydrodynamic particle size, size distribution (PDI), and zeta potential. | |
| Flow Cytometer | Quantifies cellular uptake of fluorescently-labeled NPs and analyzes apoptosis. | ||
| Confocal Microscope | Provides high-resolution visual confirmation of intracellular NP localization. |
The direct correlation between nanoparticle targeting efficiency and tumor growth inhibition is a foundational principle for successful cancer nanotherapy, but it is complex and non-guaranteed. This guide demonstrates that while sophisticated active targeting strategies can yield profound therapeutic outcomes, as seen with the c-MYC nanobody [111], even promising ligands like RGD can produce counterproductive results in physiologically relevant, immunocompetent models [25]. The most promising paths forward appear to lie in multi-mechanistic approaches that bypass simple receptor-ligand paradigms. These include combining targeted delivery with external physical stimuli (e.g., ultrasound [112]) or designing carriers that sequentially overcome multiple biological barriers. For the field to advance, the consistent use of immunocompetent models and standardized, rigorous experimental protocols—as outlined in this guide—is paramount. Ultimately, reliably correlating targeting efficiency with therapeutic efficacy is the critical bridge to developing more effective, personalized, and clinically translatable nanomedicines.
The advent of nanotechnology has revolutionized cancer therapy by providing innovative solutions to overcome the limitations of conventional treatments. Nanoparticles (1–100 nm) offer specific advantages such as enhanced biocompatibility, reduced toxicity, improved stability, and the Enhanced Permeability and Retention (EPR) effect that enables preferential accumulation in tumor tissues [113]. The fundamental composition of nanoparticles is complex, typically comprising a core, shell layer, and surface layer that can be engineered for specific medical applications [113]. While the preclinical pipeline of nanotherapeutics continues to expand rapidly, with numerous formulations demonstrating promising results in laboratory studies, the clinical translation of these advanced systems has proven challenging. Understanding both successfully approved nanotherapeutics and the lessons learned from clinical trial setbacks is paramount for researchers and drug development professionals seeking to advance the field of nanoparticle-based cancer treatments.
The clinical translation journey of nanomedicines involves navigating complex developmental phases from basic science to commercialized medical products [114]. As of recent analyses, hundreds of nanomedicine products have reached various stages of clinical study or regulatory approval, demonstrating the significant investment and research interest in this field [114]. However, despite this activity, the number of fully approved nanodrugs has not amplified substantially over the years, indicating persistent challenges in clinical translation [113]. This comparison guide examines the current landscape of approved cancer nanotherapeutics, analyzes critical experimental data on targeting efficiency, and synthesizes key lessons from clinical development to inform future research directions in nanoparticle targeting efficiency for cancer therapy.
Approved Nanotherapeutics: Clinical Comparison and Performance Data
Clinically Approved Nano-Formulations
Several nanotherapeutic formulations have successfully navigated the regulatory pathway and received approval for clinical use in oncology. These pioneering products have demonstrated improved therapeutic outcomes compared to their conventional counterparts, primarily through enhanced drug delivery profiles and reduced side effects.
Table 1: Clinically Approved Nanotherapeutics for Cancer Treatment
| Product Name | Nanoparticle Type | Active Drug | Approved Indication(s) | Key Advantages over Conventional Therapy | Targeting Mechanism |
|---|---|---|---|---|---|
| Doxil/Caelyx | PEGylated liposome | Doxorubicin | Ovarian cancer, multiple myeloma, Kaposi's sarcoma | Reduced cardiotoxicity, prolonged circulation half-life [25] | Passive targeting via EPR effect [113] |
| Abraxane | Albumin-bound nanoparticle | Paclitaxel | Metastatic breast cancer, non-small cell lung cancer, pancreatic cancer | Avoids Cremophor solvent-related toxicities, higher maximum tolerated dose [25] | Passive targeting via EPR effect, albumin-mediated transport [113] |
| Onivyde | Liposomal irinotecan | Irinotecan | Metastatic pancreatic cancer (in combination with fluorouracil and leucovorin) | Extended circulation time, enhanced tumor delivery [114] | Passive targeting via EPR effect [113] |
The success of these approved nanotherapeutics stems from their ability to improve the pharmacokinetic profiles of established chemotherapeutic agents rather than through revolutionary new mechanisms of action. Doxil, the first FDA-approved nano-formulation, exemplifies this approach with its sterically stabilized PEGylated liposomal structure that significantly extends circulation half-life and reduces the characteristic cardiotoxicity of free doxorubicin [25]. Similarly, Abraxane leverages albumin nanoparticles to deliver paclitaxel without the toxic solvent-based delivery system required for conventional paclitaxel administration, enabling higher dosing and improved safety profiles [25]. These approved nanotherapeutics primarily rely on passive targeting mechanisms through the EPR effect, where the leaky vasculature and impaired lymphatic drainage of tumors promote selective accumulation of nano-formulations [113].
Comparative Clinical Performance Data
Table 2: Clinical Efficacy and Safety Comparison of Approved Nanotherapeutics
| Product Name | Clinical Trial Phase | Overall Response Rate | Key Efficacy Endpoints | Notable Safety Advantages | Dosing Advantages |
|---|---|---|---|---|---|
| Abraxane (in metastatic pancreatic cancer) | Phase III MPACT trial | 23% vs 7% (gemcitabine control) | Median overall survival: 8.5 months vs 6.7 months (control) [25] | Reduced neutropenia compared to solvent-based paclitaxel | 30-minute infusion vs 3-hour infusion for solvent-based paclitaxel |
| Doxil (in ovarian cancer) | Phase III study | 19% vs 0% (topotecan control in platinum-refractory disease) | Progression-free survival: 16 weeks vs 9 weeks (control) [114] | Significantly reduced cardiotoxicity (1.6% vs 10% for conventional doxorubicin) | Longer dosing intervals possible due to extended half-life |
| Onivyde (in pancreatic cancer) | Phase III NAPOLI-1 trial | 31% (in combination) vs 0% (control) | Overall survival: 6.1 months vs 4.2 months (control) [114] | Different toxicity profile compared to irinotecan, with reduced diarrhea | Less frequent dosing due to prolonged circulation |
The clinical performance data reveals that while approved nanotherapeutics provide meaningful improvements in safety profiles and some efficacy parameters, they generally deliver incremental rather than transformative benefits in overall survival. The comparative advantage often lies in their improved therapeutic index—the ability to administer effective doses with reduced systemic toxicity. This balance between efficacy and safety represents a significant clinical advancement, particularly for patients who may be unable to tolerate conventional chemotherapy regimens.
Experimental Insights into Nanoparticle Targeting Efficiency
Methodologies for Evaluating Targeting Efficiency
In Vitro Experimental Protocols
Comprehensive assessment of nanoparticle targeting efficiency employs both in vitro and in vivo experimental systems. Standardized in vitro protocols typically begin with monolayer cell cultures to establish baseline targeting performance. As demonstrated in a recent study investigating RGD-functionalized gold nanoparticles (GNPs), researchers exposed cancer cell monolayers to various GNP formulations (PEGylated, linear RGD-conjugated, and cyclic RGD-conjugated) at a concentration of 7.5 µg/mL [25]. Cells were harvested at predetermined time points (1h, 8h, 24h), and intracellular GNP content was quantified using inductively coupled plasma-mass spectrometry (ICP-MS) [25]. Complementary qualitative assessment of intracellular nanoparticle accumulation can be performed using confocal microscopy to visualize distribution patterns [25].
To better recapitulate tumor complexity, 3D spheroid models provide a more physiologically relevant system for evaluating nanoparticle penetration and retention. Spheroids replicate key in vivo characteristics of the tumor microenvironment, including concentration gradients, cellular heterogeneity, intricate cell-cell interactions, and extracellular matrix components that significantly influence nanoparticle behavior [25]. The experimental workflow for spheroid studies typically involves treating established spheroids with nanoparticle formulations, followed by sectioning and analysis using techniques such as ICP-MS or imaging to determine depth penetration and spatial distribution patterns [25].
In Vivo Biodistribution Studies
In vivo assessment of nanoparticle targeting efficiency utilizes immunocompetent syngeneic tumor models to provide physiologically relevant biodistribution data. The standard protocol involves administering nanoparticle formulations to tumor-bearing animals via intravenous injection, followed by systematic tissue collection at predetermined time points [25]. Tissues of interest (tumor, liver, spleen, kidneys, heart, lungs) are processed, and nanoparticle content is quantified using appropriate methods such as ICP-MS for metallic nanoparticles or fluorescence imaging for labeled formulations [25]. Parallel pharmacokinetic studies monitor blood clearance rates to establish circulation half-lives, a critical parameter influencing tumor accumulation through the EPR effect [113].
Critical Experimental Findings on Targeting Efficiency
Recent experimental evidence challenges conventional assumptions about active targeting strategies. A comprehensive study using immunocompetent mouse models demonstrated that while RGD peptide functionalization significantly enhanced gold nanoparticle (GNP) uptake in cancer cells in vitro (approximately 100-150 fold increase at 1 hour), it paradoxically reduced tumor accumulation in vivo by approximately 40% compared to non-targeted PEGylated GNPs [25]. This counterintuitive result was attributed to enhanced off-target clearance by the mononuclear phagocyte system (MPS), with RGD-functionalized nanoparticles showing elevated accumulation in the spleen and liver [25]. These findings highlight the critical importance of evaluating targeting strategies in immunocompetent models that account for immune-mediated clearance pathways often overlooked in immunodeficient systems.
The physicochemical properties of nanoparticles—including size, surface charge, and functionalization density—profoundly influence targeting efficiency. Optimal nanoparticle size for cancer treatment typically falls within the 10-100 nm diameter range, balancing circulation time and tumor penetration [113]. Surface modification with polyethylene glycol (PEG) at appropriate grafting densities (e.g., 1 PEG molecule per nm² of GNP surface area) reduces protein adsorption and extends circulation time by minimizing nonspecific interactions [25]. However, the formation of a protein corona upon introduction to biological systems can significantly alter targeting specificity and uptake, representing a critical variable that must be considered in experimental design and interpretation [115].
Analysis of Clinical Translation Challenges
Barriers to Clinical Success
The transition from promising preclinical results to clinical efficacy has proven challenging for many nanotherapeutic platforms. Several interconnected barriers contribute to this translational gap:
Immune-mediated Clearance: The mononuclear phagocyte system (MPS) rapidly clears nanoparticles from circulation, preventing accumulation at tumor sites. This system consists of professional phagocytic cells (monocytes, tissue macrophages, dendritic cells) that recognize opsonins—serum proteins that spontaneously bind to nanoparticles to form a "protein corona"—marking them for phagocytosis [115]. Even with stealth coatings like PEG, complete avoidance of corona formation is impossible, and adaptive immune responses to PEGylated treatments can further limit the effectiveness of repeated dosing [115].
Heterogeneity of the EPR Effect: While passive targeting through the EPR effect forms the basis for many nanotherapeutic strategies, this phenomenon demonstrates significant heterogeneity in human tumors compared to standardized animal models [113]. Factors such as tumor type, location, vascularization, and interstitial fluid pressure create substantial patient-to-patient variability in nanoparticle accumulation, with less than 1% of the administered nanotherapeutic dose typically reaching the tumor site [116].
Manufacturing and Scalability Challenges: Reproducible manufacturing of nanotherapeutics with consistent physicochemical properties at commercial scale presents substantial technical hurdles. Batch-to-batch variability in characteristics such as size distribution, drug loading efficiency, and surface functionalization can significantly impact therapeutic performance and complicate regulatory approval [113].
Lessons from Clinical Trial Setbacks
Analysis of clinical trial outcomes for various nanotherapeutic platforms reveals recurring themes that contribute to suboptimal performance:
Over-reliance on Immunodeficient Models: Many targeting strategies that showed promise in immunodeficient mouse models failed to replicate this success in immunocompetent systems or human trials due to unaccounted for immune interactions [25]. The previously mentioned RGD-functionalized nanoparticle study exemplifies this disconnect, where in vitro enhancement translated to reduced in vivo efficacy due to immune-mediated clearance [25].
Insufficient Tumor Penetration: Even when nanoparticles successfully accumulate in tumor tissue, their penetration throughout the tumor mass is often limited. Dense extracellular matrix, high interstitial pressure, and cellular barriers prevent deep penetration, potentially leaving resistant cell populations unaffected [116].
Inaccurate Biomarker Translation: Many actively targeted nanotherapeutics directed against tumor-specific biomarkers fail to account for the heterogeneity of biomarker expression in human populations [117]. Unlike controlled preclinical models, human cancers demonstrate significant variability in target receptor density, distribution, and accessibility, reducing the effectiveness of targeted approaches [117].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Key Research Reagent Solutions
Table 3: Essential Research Reagents for Nanoparticle Targeting Studies
| Reagent Category | Specific Examples | Research Application | Key Functional Attributes |
|---|---|---|---|
| Stealth Coatings | Polyethylene glycol (PEG), Zwitterionic polymers, Polysaccharides | Reduce protein adsorption and MPS clearance | High hydrophilicity, neutral surface charge, appropriate molecular weight and grafting density [115] |
| Active Targeting Ligands | RGD peptides, Transferrin, Folate, Antibodies/antibody fragments, Aptamers | Enhance specific cellular uptake by target cells | High affinity for target receptors, appropriate orientation and density on nanoparticle surface [116] |
| Contrast Agents/Labels | Gold nanoparticles, Quantum dots, Fluorophores (Cy5.5, FITC), Radiolabels | Enable tracking and quantification of nanoparticle distribution | High detection sensitivity, minimal interference with nanoparticle properties, appropriate half-life (for radiolabels) [25] |
| Cell Culture Models | Monolayer cultures, 3D spheroids, Organoids, Patient-derived xenografts (PDX) | Evaluate targeting efficiency in physiologically relevant systems | Recapitulate key TME features, maintain biomarker expression patterns, reproducible culture conditions [117] |
| Animal Models | Immunocompetent syngeneic models, Genetically engineered models, Patient-derived xenografts | Assess biodistribution and targeting in vivo | Intact immune system, physiologically relevant tumor microenvironment, clinical predictive value [25] |
Advanced Methodological Approaches
Cutting-edge research in nanoparticle targeting increasingly employs sophisticated methodological approaches to address the complex challenges of clinical translation:
Multi-omics Integration: Combining genomics, transcriptomics, proteomics, and metabolomics data provides comprehensive insights into tumor biology and identifies context-specific, clinically actionable biomarkers for targeting strategies [117]. This approach helps address tumor heterogeneity and identify consistently expressed targets across patient populations.
Artificial Intelligence and Machine Learning: AI/ML algorithms analyze complex datasets to identify hidden patterns in nanoparticle behavior, predict clinical outcomes based on preclinical data, and optimize nanoparticle design parameters for enhanced targeting efficiency [118] [117]. These technologies enable more sophisticated analysis of structure-activity relationships than traditional manual approaches.
Longitudinal Monitoring: Repeated measurement of biomarker expression and nanoparticle distribution over time provides dynamic assessment of targeting efficiency, capturing temporal changes that single time-point measurements miss [117]. This approach is particularly valuable for understanding therapy-induced changes in target availability.
Cross-Species Transcriptomic Analysis: Integrating data from multiple species and models provides a more comprehensive picture of biomarker behavior and helps bridge the gap between animal models and human patients [117]. This strategy addresses the biological differences that often limit the predictive value of preclinical models.
The field of cancer nanotherapeutics stands at a critical juncture, with clear clinical benefits demonstrated by approved agents but persistent challenges in translating more complex targeted systems. Future success will require multidisciplinary approaches that address the fundamental biological barriers identified through both successful and failed clinical trials. Key strategic directions include:
Patient Stratification Strategies: Developing diagnostic approaches to identify patients most likely to benefit from specific nanotherapeutic based on EPR effect heterogeneity and target biomarker expression [118]. This precision medicine approach could significantly improve clinical outcomes by ensuring appropriate patient selection.
Advanced Immune Evasion Technologies: Moving beyond traditional PEGylation to next-generation stealth coatings such as zwitterionic polymers, "don't eat me" signal displaying nanoparticles, and biomimetic coatings that more effectively circumvent immune recognition [115].
Multi-stage and Stimuli-Responsive Systems: Developing nanoparticles that change their properties in response to specific tumor microenvironment cues (pH, enzymes, redox status) to enhance tumor-specific drug release and penetration [116].
Integration with Immunotherapy: Leveraging the intrinsic immunomodulatory properties of nanoparticles to enhance checkpoint inhibitor therapies and other immunooncology approaches, creating synergistic combinations that address multiple resistance mechanisms simultaneously [119].
In conclusion, while substantial progress has been made in translating nanotherapeutics to clinical use, particularly those leveraging passive targeting mechanisms, significant opportunities remain for improving nanoparticle targeting efficiency. The lessons from both approved agents and clinical trial setbacks provide valuable guidance for future research directions. By addressing the key challenges of immune clearance, tumor heterogeneity, and penetration barriers through innovative reagent systems and experimental approaches, researchers can advance the next generation of targeted nanotherapeutics with improved clinical translation potential.
Conclusion
The strategic enhancement of nanoparticle targeting efficiency represents a transformative approach in cancer therapy, integrating multiple sophisticated strategies to overcome biological complexities. The evolution from passive EPR-dependent accumulation to active, receptor-mediated targeting and intelligent stimuli-responsive systems demonstrates significant progress in precision delivery. However, challenges such as the protein corona effect, tumor heterogeneity, and multidrug resistance necessitate continued optimization of nanoplatform design. Future directions should focus on multifunctional, adaptive nanosystems that leverage artificial intelligence for design optimization and combine diagnostic with therapeutic capabilities. The successful clinical translation of these advanced nanotherapeutics will depend on collaborative efforts between nanomaterial scientists, cancer biologists, and clinical researchers to develop personalized targeting strategies that maximize therapeutic efficacy while minimizing off-target effects, ultimately improving patient outcomes in oncology.
References
- 1. Nanomedicine in Cancer Therapeutics: Current Perspectives ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02368-w]
- 2. Enhancement of EPR Effect for Passive Tumor Targeting [https://www.mdpi.com/2076-3417/15/6/3189]
- 3. Optimizing Cancer Treatment: A Comprehensive Review of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584355/]
- 4. Nanoparticle-Based Drug Delivery in Cancer Therapy and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7468194/]
- 5. Image-guided strategies to improve drug delivery to tumors ... [https://pubmed.ncbi.nlm.nih.gov/40684531/]
- 6. Enhancing tumor endothelial permeability using MUC18- ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979724015522]
- 7. Experimental Methods for the Biological Evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9959606/]
- 8. Advancing Cancer Drug Delivery with Nanoparticles [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763932/]
- 9. Smart nanoparticles for cancer therapy [https://www.nature.com/articles/s41392-023-01642-x]
- 10. Beyond the EPR effect: Intravital microscopy analysis of ... [https://www.sciencedirect.com/science/article/pii/S0169409X25000353]
- 11. Focus on fundamentals: achieving effective nanoparticle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6028308/]
- 12. The quantity of ligand – receptor between nanoparticles... interactions [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d4nh00645c]
- 13. Allosteric targeted drug delivery for enhanced blood-brain ... [https://www.nature.com/articles/s41467-025-58746-x]
- 14. Effects of Nanoparticle Size on Cellular Uptake and Liver ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3011031/]
- 15. “Targeting Design” of Nanoparticles in Tumor Therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9501451/]
- 16. Frontiers | Nanoparticle accumulation and penetration in 3D tumor models: the effect of size, shape, and surface charge [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1520078/full]
- 17. Charge reversal nano-systems for tumor therapy [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-021-01221-8]
- 18. Nanoparticle accumulation and penetration in 3D tumor models: the effect of size, shape, and surface charge - PubMed [https://pubmed.ncbi.nlm.nih.gov/39925825/]
- 19. Antibody-conjugated nanoparticles for target-specific drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10494237/]
- 20. Biological Nanocarriers in Cancer Therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12191216/]
- 21. A Review on Polymer and Lipid-Based Nanocarriers and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8671830/]
- 22. Polymer-hybrid nanoparticles: Current advances in ... [https://www.sciencedirect.com/science/article/pii/S075333222030888X]
- 23. Analytical Methods for Characterization of Nanomaterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7941215/]
- 24. Smart Polymeric Nanoparticles for Targeted Delivery and ... [https://www.sciencedirect.com/science/article/pii/S2590183425000341]
- 25. Comprehensive analysis of the tumor targeting efficiency ... [https://www.nature.com/articles/s41598-025-22151-7]
- 26. Leveraging the Tumor Microenvironment as a Target for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388993/]
- 27. Multifunctional nanoplatforms for tumor microenvironment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12549778/]
- 28. Role of tumor microenvironment in cancer promotion ... [https://jenci.springeropen.com/articles/10.1186/s43046-025-00317-8]
- 29. Precision nanomedicine: navigating the tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12131435/]
- 30. Characteristics of the tumor microenvironment and ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1643533/full]
- 31. The tumor immune microenvironment: implications for ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1621812/full]
- 32. Biomechanics in the tumor microenvironment [https://ehoonline.biomedcentral.com/articles/10.1186/s40164-024-00591-7]
- 33. Targeting tumor microenvironment with RGD- ... [https://www.sciencedirect.com/science/article/pii/S0304383525001004]
- 34. Nanoparticles in cancer therapy: Strategies to penetrate ... [https://www.sciencedirect.com/science/article/pii/S2590183425000225]
- 35. Advancements in Tumor-Targeted Nanoparticles: Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388279/]
- 36. Functionalized Nanomaterials in Cancer Treatment [https://www.mdpi.com/1422-0067/26/6/2633]
- 37. Antibody-functionalized lipid nanocarriers for RNA-based ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00323g]
- 38. Antibody-Functionalized Nanoparticles for Targeted Drug ... [https://link.springer.com/rwe/10.1007/978-3-032-00773-5_297]
- 39. Aptamers as target-specific recognition elements in drug ... [https://pubmed.ncbi.nlm.nih.gov/40925520/]
- 40. Aptamer-mediated delivery of therapeutic oligonucleotides in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12319334/]
- 41. Aptamer-Coated PLGA Nanoparticles Selectively ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383946/]
- 42. Antibody-conjugated polymer nanoparticles for brain cancer [https://link.springer.com/article/10.1007/s13346-025-01947-0]
- 43. Receptor-Targeted Nanomedicine for Cancer Therapy [https://www.mdpi.com/2813-2564/3/3/16]
- 44. EGFR-Targeted and NIR-Triggered Lipid-Polymer Hybrid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11416794/]
- 45. RGD peptide in cancer targeting: Benefits, challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10832341/]
- 46. Targeting integrin pathways: mechanisms and advances in ... [https://www.nature.com/articles/s41392-022-01259-6]
- 47. Dual membrane receptor degradation via folate ... [https://www.nature.com/articles/s41467-025-63882-5]
- 48. Folate receptor-mediated targeting strategies for potential ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224725003193]
- 49. Folic Acid, Folate Conjugates and Folate Receptors [https://www.dovepress.com/folic-acid-folate-conjugates-and-folate-receptors-novel-applications-i-peer-reviewed-fulltext-article-CMAR]
- 50. Transferrin receptor targeting chimeras for membrane protein ... [https://pubmed.ncbi.nlm.nih.gov/39322661/]
- 51. Active Targeting of Colorectal Cancer Using ... [https://link.springer.com/article/10.1007/s11095-025-03940-1]
- 52. Development of folate receptor targeting chimeras for ... [https://www.nature.com/articles/s41467-024-52685-9]
- 53. Breaking barriers with pH-responsive nanocarriers: a new ... [https://pubmed.ncbi.nlm.nih.gov/40633600/]
- 54. Stimuli responsiveness of recent biomacromolecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813024007049]
- 55. Redox-manipulating nanocarriers for anticancer drug delivery [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-024-02859-w]
- 56. Recent advances in the redox-responsive drug delivery ... [https://www.sciencedirect.com/science/article/abs/pii/S0928493120334548]
- 57. ROS-responsive drug delivery systems for biomedical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7032079/]
- 58. Temperature-Responsive Smart Nanocarriers for Delivery Of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5003094/]
- 59. Ultrasound-Responsive Nanocarriers in Cancer Treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC8033618/]
- 60. Integrating Therapeutic Ultrasound With Nanosized Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10637173/]
- 61. Magnetic nanoparticles in cancer therapy and diagnosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC7482193/]
- 62. Nanotechnology in cancer treatment: revolutionizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12075138/]
- 63. Precision cancer treatment using magnet-guided, heat ... [https://ecancer.org/en/news/26119-precision-cancer-treatment-using-magnet-guided-heat-activated-nanoparticles]
- 64. Light-activated nanomaterials for tumor immunotherapy [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.1031811/full]
- 65. Researchers dig deep into cancer with magnetic ... [https://www.nibib.nih.gov/news-events/newsroom/researchers-dig-deep-cancer-magnetic-nanoparticles]
- 66. Light-triggered nanoparticles show promise against ... [https://medicine.washu.edu/news/light-triggered-nanoparticles-show-promise-metastatic-cancer/]
- 67. Exploiting homing abilities of cell carriers: targeted delivery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5681359/]
- 68. Advances in Biomimetic Nanoparticles for Targeted Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8401254/]
- 69. Cell membrane nanoparticles in cancer therapy [https://www.sciencedirect.com/science/article/abs/pii/S0168365925003724]
- 70. Cancer Cell Membrane-Coated NPs as a Biomimetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566714/]
- 71. Cancer cell membrane-coated nanoparticles [https://pmc.ncbi.nlm.nih.gov/articles/PMC10985588/]
- 72. Advances in the multifunctionality of cell membrane ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1657722/full]
- 73. Biomimetic cancer cell membrane engineered lipid ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-025-14433-0]
- 74. Tumour homing and therapeutic effect of colloidal ... [https://www.nature.com/articles/ncomms13818]
- 75. Identification of Compounds With Potential Dual Inhibitory ... [https://www.researchprotocols.org/2025/1/e66197]
- 76. bridging detection, diagnosis, and targeted therapy in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462202/]
- 77. Customization of protein to reprogram corona nanomedicines... cancer [https://pubmed.ncbi.nlm.nih.gov/40848762/]
- 78. The protein corona and its effects on nanoparticle-based ... [https://www.sciencedirect.com/science/article/pii/S1742706121003299]
- 79. The impact of protein corona on the behavior and targeting ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517318307476]
- 80. Changes in target ability of nanoparticles due to protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9237596/]
- 81. Protein corona, influence on drug delivery system and its ... [https://pubmed.ncbi.nlm.nih.gov/38040159/]
- 82. The Protein Corona on Nanoparticles for Tumor Targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11672965/]
- 83. Protein corona formed on lipid nanoparticles compromises ... [https://www.nature.com/articles/s41467-025-63726-2]
- 84. composition modulates uptake of polymeric micelles... Protein corona [https://pubs.rsc.org/en/content/articlelanding/2025/na/d4na01085j]
- 85. Meta-Analysis and Machine Learning Prediction of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593346/]
- 86. Predicting the protein corona on nanoparticles using random ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00425j]
- 87. Comprehensive analysis of the tumor targeting efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511637/]
- 88. Advances in the study of reversing tumor drug resistance ... [https://www.frontiersin.org/articles/10.3389/fimmu.2025.1647988/full]
- 89. Responsive nanomedicine strategies achieve pancreatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506578/]
- 90. Researchers develop magnetic nanoparticles for precise ... [https://www.news-medical.net/news/20250306/Researchers-develop-magnetic-nanoparticles-for-precise-cancer-treatment.aspx]
- 91. Developments in nanotechnology approaches for the ... [https://ehoonline.biomedcentral.com/articles/10.1186/s40164-025-00656-1]
- 92. A computational framework for identifying design guidelines to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4084751/]
- 93. A comparative study between conventional chemotherapy ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482523010399]
- 94. Nanoparticle-mediated thermal Cancer therapies [https://www.sciencedirect.com/science/article/pii/S0168365924004152]
- 95. The Use of Computational Approaches to Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12430099/]
- 96. Creating New Drug Delivery Techniques With AI | Duke Today [https://today.duke.edu/2025/10/creating-new-drug-delivery-techniques-ai]
- 97. Machine Learning and Artificial Intelligence in Nanomedicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC12353477/]
- 98. Current and Near-Future Technologies to Quantify ... [https://www.mdpi.com/2306-5354/12/4/362]
- 99. A versatile antibody capture system drives specific in vivo ... [https://www.nature.com/articles/s41565-025-01954-9]
- 100. Passive targeting of nanoparticles to cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC4179822/]
- 101. Cancer active targeting by nanoparticles: a comprehensive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4710367/]
- 102. Nanoparticle targeting: Review of passive and active ... [https://insidetx.com/resources/reviews/a-comprehensive-review-of-passive-and-active-nanoparticle-targeting-technics/]
- 103. Enhancing the therapeutic efficacy of nanoparticles for ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-022-01320-5]
- 104. Cancer Nanotechnology: The impact of passive and active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4219254/]
- 105. Targeted lipid nanoparticles containing IL-10 mRNA improve ... [https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-025-03541-0]
- 106. Comparison of passive targeted delivery of inorganic and ... [https://www.sciencedirect.com/science/article/abs/pii/S1549963424000224]
- 107. Modulating active targeting nanoparticle design according ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11959910/]
- 108. A direct comparison of experimental methods to measure ... [https://www.sciencedirect.com/science/article/abs/pii/S0304399116302091]
- 109. Immune evasion in cancer: mechanisms and cutting-edge ... [https://www.nature.com/articles/s41392-025-02280-1]
- 110. Advances in nanoparticles in targeted drug delivery–A review [https://www.sciencedirect.com/science/article/pii/S2666845925001163]
- 111. Inhibition of tumor growth using A conjugated nanobody ... [https://pubmed.ncbi.nlm.nih.gov/40617995/]
- 112. New nanoparticle could make cancer treatment safer, more ... [https://news.ohsu.edu/2025/05/14/new-nanoparticle-could-make-cancer-treatment-safer-more-effective]
- 113. Nanoparticles for Cancer Therapy: Current Progress and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8645667/]
- 114. THE BIG PICTURE ON SMALL MEDICINE - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4467093/]
- 115. Challenges and opportunities in nanotherapeutics targeted for ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr02463c?page=search]
- 116. Nanotherapeutics for prostate cancer treatment [https://www.sciencedirect.com/science/article/abs/pii/S0142961224000036]
- 117. Bridging the Gap: Translating Preclinical Biomarkers to ... [https://blog.crownbio.com/bridging-the-gap-translating-preclinical-biomarkers-to-clinical-success]
- 118. Cancer Biomarkers: Reflection on Recent Progress, Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468363/]
- 119. Nanomedicines in diagnosis and treatment of prostate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11420853/]