Lipid Nanoparticles vs. Viral Vectors: A Strategic Guide to Gene Delivery Technologies
This article provides a comprehensive comparison of lipid nanoparticle (LNP) and viral vector platforms for gene delivery, tailored for researchers and drug development professionals.
Lipid Nanoparticles vs. Viral Vectors: A Strategic Guide to Gene Delivery Technologies
Abstract
This article provides a comprehensive comparison of lipid nanoparticle (LNP) and viral vector platforms for gene delivery, tailored for researchers and drug development professionals. It covers foundational mechanisms, current clinical applications across various diseases, strategies to overcome technical and immunological challenges, and a direct comparative analysis of safety, efficacy, and scalability. The scope extends from basic principles to advanced optimization techniques and future directions, serving as a strategic resource for selecting the appropriate delivery system for specific therapeutic goals.
Core Principles and Evolutionary Paths of Gene Delivery Systems
The success of gene therapy hinges on the efficient delivery of genetic cargo into target cells, a process facilitated by engineered vectors. These platforms are broadly categorized into viral and non-viral systems. Viral vectors, including adeno-associated virus (AAV), lentivirus (LV), and adenovirus (Ad), are engineered from naturally evolving viruses and leverage their innate ability to infect cells [1]. In contrast, non-viral vectors such as lipid nanoparticles (LNPs) are synthetic constructs designed to overcome the limitations of their viral counterparts [2]. The choice between these platforms involves a critical trade-off between delivery efficiency, cargo capacity, safety profile, and manufacturability [3] [4]. Viral vectors generally offer high transduction efficiency and long-lasting gene expression, while LNPs provide a safer profile with lower immunogenicity, larger cargo capacity, and the significant advantage of being suitable for re-dosing [5]. This guide provides a objective comparison of these platforms, focusing on their mechanisms, performance data, and experimental applications for researchers and drug development professionals.
Platform Mechanisms and Workflows
Viral Vector Mechanisms
Viral vectors are modified viruses that have been rendered replication-incompetent but retain their ability to deliver genetic material into cells. Their mechanisms are distinguished by their routes of cell entry and genomic handling.
- Adeno-Associated Virus (AAV): AAV vectors bind to cell surface primary receptors (e.g., HSPG) and coreceptors prior to clathrin-mediated endocytosis. The low pH of the endosome triggers conformational changes in the viral capsid, leading to endosomal escape. The single-stranded DNA genome is then released into the nucleus, where it predominantly remains as a non-integrating episome, facilitating long-term transgene expression in non-dividing cells [1] [6].
- Lentivirus (LV): As a complex retrovirus, LV enters cells via envelope-mediated fusion, often pseudotyped with the VSV-G protein. Following entry, the viral RNA genome is reverse-transcribed into double-stranded DNA in the cytoplasm. This DNA, along with viral integrase, is imported into the nucleus and integrates into the host cell genome, enabling stable, long-term transgene expression that is passed on to daughter cells [1] [7].
- Adenovirus (Ad): Ad vectors bind to the coxsackievirus and adenovirus receptor (CAR) and are internalized via endocytosis. The viral capsid is destabilized in the endosome, and the DNA genome is transported to the nucleus where it remains episomal. Ad vectors can efficiently transduce both dividing and non-dividing cells and typically elicit high levels of transient gene expression, but also strong immune responses [1] [6].
LNP Mechanism and Workflow
LNPs are synthetic, multi-component systems that self-assemble into nanoparticles to encapsulate and protect nucleic acids. The typical LNP formulation for mRNA delivery consists of four key lipids: an ionizable cationic lipid, a phospholipid, cholesterol, and a PEG-lipid [2] [8].
The delivery mechanism involves a sequence of critical steps, visualized in the workflow below.
Figure 1. LNP Delivery Workflow. The process begins with systemic administration. Following cellular uptake via endocytosis, the key mechanistic step is endosomal escape: the acidic environment of the endosome protonates the ionizable cationic lipids, promoting inverted hexagonal phase structure formation and fusion with the endosomal membrane, resulting in cytoplasmic release of the nucleic acid payload [2] [6]. The mRNA is then translated into the therapeutic protein, resulting in transient expression.
Comparative Performance Data
The following tables summarize the key characteristics and performance metrics of AAV, Lentivirus, Adenovirus, and LNP platforms, based on current literature and clinical data.
Table 1. Comparative Overview of Gene Delivery Platforms
| Feature | AAV | Lentivirus (LV) | Adenovirus (Ad) | Lipid Nanoparticles (LNP) |
|---|---|---|---|---|
| Payload Type | ssDNA | RNA (reverse transcribed to dsDNA) | dsDNA | mRNA, siRNA, plasmid DNA, proteins [5] |
| Cargo Capacity | <5 kb [5] [4] | ~8 kb [7] | ~8-36 kb [1] | >10 kb, versatile/unrestricted [5] [4] |
| Integration Profile | Predominantly episomal (<0.1% integrates) [1] | Integrates into host genome [1] | Episomal | Non-integrating [3] |
| Expression Duration | Long-term (months to years) [3] | Long-term (stable in dividing cells) [7] | Short-term (transient) [1] | Short-term (transient) [3] |
| Immunogenicity | Moderate; pre-existing immunity concerns [1] | Moderate; ex vivo use minimizes this [7] | High; strong inflammatory response [1] [6] | Low; suitable for re-dosing [3] [5] |
| Primary Applications | Inherited retinal diseases, SMA, hemophilia [1] [9] | Ex vivo cell therapy (CAR-T, HSPCs) [1] [7] | Vaccines, oncolytic therapy [1] [7] | mRNA vaccines, siRNA therapeutics, gene editing [3] [9] |
Table 2. Experimental and Manufacturing Considerations
| Consideration | AAV | Lentivirus (LV) | Adenovirus (Ad) | Lipid Nanoparticles (LNP) |
|---|---|---|---|---|
| Titer/ Potency (Typical) | High (in vivo) [3] | High (ex vivo) [3] [7] | Very High [1] | Moderate to High (cargo and cell-type dependent) [3] [2] |
| In Vivo Delivery Efficiency | High for specific tissues (e.g., liver, muscle, CNS) [3] | Developing for in vivo use [7] | High for multiple tissues [1] | High for liver; improving for extrahepatic targets [5] [4] |
| Manufacturing Complexity | High; biological production, difficult purification [4] | High; multi-plasmid transfection of cells [7] | Moderate [1] | Low; rapid, scalable synthetic process [3] [4] |
| Relative Cost of Goods (COGS) | Very High [4] | High [7] | Moderate | Low [4] |
Key Experimental Protocols
Protocol: In Vivo Gene Editing with LNPs
This protocol details the use of LNPs to deliver CRISPR-Cas9 components for gene editing in mouse liver, a common application for this platform [10] [2].
- LNP Formulation: Utilize a microfluidic device to mix an ethanolic lipid solution (containing ionizable lipid, DSPC, cholesterol, and DMG-PEG2000 at a defined molar ratio) with an aqueous solution containing CRISPR-Cas9 mRNA and sgRNA at a 1:3 volumetric flow rate ratio [2].
- Dialysis and Concentration: Dialyze the formed LNP suspension against a suitable buffer (e.g., PBS, pH 7.4) for several hours to remove ethanol and exchange the external buffer. Subsequently, concentrate the LNPs using centrifugal filter units.
- Characterization: Measure the particle size, polydispersity index (PDI), and zeta potential of the LNPs using dynamic light scattering. Determine encapsulation efficiency using a Ribogreen assay.
- In Vivo Administration: Administer the LNP formulation to mice via systemic tail-vein injection at a dosage of 0.5-3 mg mRNA per kg body weight. A common dose for liver editing is 1 mg/kg [10].
- Analysis: After 3-7 days, harvest target tissues (e.g., liver). Extract genomic DNA and assess editing efficiency at the target locus using next-generation sequencing (NGS) or T7 Endonuclease I (T7EI) assay. Evaluate potential off-target editing at predicted sites.
Protocol: Directed Evolution of Engineered VLPs
This advanced protocol, adapted from a recent Nature Biotechnology study, uses barcoded sgRNAs to evolve engineered virus-like particles (eVLP) with improved properties, showcasing a high-throughput viral vector engineering method [10].
- Library Construction: Generate a library of eVLP production vectors, each encoding a unique capsid variant and a corresponding uniquely barcoded sgRNA.
- eVLP Production and Selection: Transfect the library into producer cells under single-variant conditions to generate a library of barcoded eVLPs. Subject this eVLP library to a selection pressure for a desired property (e.g., resistance to human serum, enhanced transduction of a specific cell type).
- Barcode Sequencing and Analysis: Transduce target cells with the post-selection eVLPs. Recover the genomic DNA from transduced cells and amplify the barcode regions by PCR. Identify enriched barcodes in the post-selection population compared to the input library via next-generation sequencing.
- Hit Validation: Re-constitute the eVLP capsid sequences corresponding to the enriched barcodes. Produce these individual eVLP hits and validate their improved performance (e.g., increased transduction efficiency, improved production titer) in secondary functional assays.
The Scientist's Toolkit: Research Reagent Solutions
Table 3. Essential Reagents for Gene Delivery Research
| Reagent / Solution | Function / Application | Key Characteristics |
|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs for nucleic acid encapsulation and endosomal escape [2] [6] | Positively charged at low pH (facilitates RNA complexation), neutral at physiological pH (reduces toxicity). Examples: DLin-MC3-DMA, SM-102. |
| PEG-Lipids | Stabilizes LNP surface; modulates pharmacokinetics and cellular tropism [2] | Prevents nanoparticle aggregation and non-specific protein adsorption. Can contribute to the "accelerated blood clearance" phenomenon upon repeated dosing [2]. |
| VSV-G Envelope Glycoprotein | Common pseudotyping protein for Lentiviral and other viral vectors [10] [7] | Confers broad tropism by binding to ubiquitous LDL receptors. Enhances vector stability and enables concentration by ultracentrifugation. |
| Polyethylenimine (PEI) | Cationic polymer for non-viral transfection, often used in research [8] | High positive charge density allows for strong DNA condensation and proton-sponge effect for endosomal escape. Can be cytotoxic. |
| Barcoded sgRNA Libraries | High-throughput screening of delivery vehicle function and evolution [10] | Allows for multiplexed tracking of vector variants in a pool. Enables directed evolution of novel capsids and selection for specific tissue tropisms. |
| GalNAc Ligands | Conjugate for targeted delivery of RNA therapeutics to hepatocytes [9] | Binds specifically to the asialoglycoprotein receptor (ASGPR) on liver cells. Enables subcutaneous administration and potent, selective liver gene silencing. |
The landscape of gene delivery is diverse, with no single platform serving as a universal solution. The choice between AAV, lentivirus, adenovirus, and LNP is dictated by the specific therapeutic goal. AAV remains the gold standard for long-term gene expression in non-dividing cells, while lentivirus is indispensable for stable genetic modification in dividing cells, especially ex vivo. Adenovirus is powerful for vaccines and oncolytic therapy where transient, high-level expression is desired. LNPs have emerged as a versatile, safe, and scalable platform, particularly suited for transient applications like mRNA vaccines and gene editing, with a superior safety profile for re-dosing [3] [4] [9]. Future progress will likely involve combining the strengths of different platforms and further engineering to overcome remaining barriers, such as pre-existing immunity to viral vectors and the targeted delivery of LNPs beyond the liver [5] [7].
Gene therapy represents a transformative approach in modern medicine, aiming to treat diseases by delivering therapeutic genetic material into a patient's cells. The success of this intervention hinges entirely on the delivery vehicle, or vector, which must safely and efficiently transport its cargo to the target cells. For decades, viral vectors dominated this landscape, leveraging the innate ability of viruses to infect cells. However, the past few years have witnessed a significant shift with the rapid ascent of non-viral vectors, particularly lipid nanoparticles (LNPs), which offer a safer and more versatile alternative [9]. This guide provides an objective comparison of these two technologies, tracing their historical evolution and providing a detailed, data-driven analysis of their current capabilities for researchers and drug development professionals.
A Tale of Two Technologies: Historical Progression
The Era of Viral Vectors
The use of viral vectors in gene therapy has a history dating back to the 1970s, with the first recorded death attributed to a viral vector administration occurring in 1999 [11]. The powerful natural infection mechanism of viruses made them an obvious candidate for early gene therapy research. Among the most prominent viral vectors are:
- Adeno-associated viruses (AAVs): Known for their favorable safety profile, as they are non-pathogenic and generally non-integrative. This has made them a preferred choice for many in vivo gene therapies, with about 72% of cell and gene therapy (CGT) trials using adenovirus or AAVs [11].
- Lentiviruses (LV): Widely used in ex vivo therapies, such as CAR-T cell treatments for blood cancers, because they can integrate their genetic material into the host genome, allowing for long-term gene expression [3] [12].
- Adenoviruses (Ad): Despite early setbacks, they remain relevant but are associated with stronger immune responses [11].
The first major milestones for viral vectors came with regulatory approvals. Spark Therapeutics' Luxturna (voretigene neparvovec) for an inherited retinal dystrophy was approved in 2017, followed by beti-cel for beta-thalassemia in 2022 [11]. To date, 29 viral vector-based gene therapies have gained market approval [12].
The Rise of Non-Viral LNPs
While viral vectors were advancing, research into non-viral methods persisted, seeking to overcome the limitations of immunogenicity and manufacturing complexity. The breakthrough for lipid nanoparticles (LNPs) came largely from their successful deployment in mRNA-based COVID-19 vaccines, which demonstrated their efficacy and safety on a global scale [3] [13].
LNPs are tiny, spherical carriers composed of lipids that encapsulate therapeutic genetic material. Their rise signifies a move towards safer, more scalable, and more versatile delivery systems [3] [13]. The first LNP-based gene therapy, Onpattro (patisiran), was approved in 2018 for the treatment of hereditary transthyretin-mediated amyloidosis [12]. A significant recent advancement is the development of the first safe and effective DNA-loaded LNPs (DNA-LNPs), which overcome the previous fatal barrier of severe immune activation and open doors for treatments for chronic diseases [14].
Head-to-Head Comparison: Performance Data
The choice between viral vectors and LNPs is multifaceted. The following tables summarize key quantitative and qualitative differences to inform research and development strategies.
Table 1: Quantitative Comparison of Key Performance Indicators
| Parameter | Viral Vectors (e.g., AAV, LV) | Lipid Nanoparticles (LNPs) |
|---|---|---|
| Transfection Efficiency | High (evolved cellular entry mechanisms) [13] | Comparable and improving, especially in specific cell lines [13] |
| Duration of Expression | Long-term or permanent (e.g., AAVs: sustained; LVs: integrated) [3] | Transient (hours to days for mRNA); longer with DNA-LNPs (~6 months) [3] [14] |
| Immunogenicity | High; pre-existing immunity and immune responses limit re-dosing [3] [11] | Lower; more suitable for repeated dosing [3] [13] |
| Scalability & Manufacturing | Complex, time-consuming, costly (CAGR 21.65% for mfg. market) [15] [13] | Relatively easy to scale; commercially viable [3] [13] |
| Payload Capacity | Limited (<~5 kb for AAV) [3] | Versatile; can deliver mRNA, siRNA, DNA, CRISPR components [3] |
| Tumor Accumulation | Varies by serotype and engineering | <10% with passive targeting; up to 89% with AI-optimized active targeting [16] |
Table 2: Qualitative Comparison of Advantages and Limitations
| Aspect | Viral Vectors | Lipid Nanoparticles |
|---|---|---|
| Key Advantages | • High-efficiency transduction• Long-term gene expression• Proven clinical success (29 approved therapies) [12] | • Low immunogenicity• Large payload capacity• Re-dosing possible• Simplified manufacturing [3] [13] |
| Key Limitations & Risks | • Immune response & toxicity (e.g., liver failure) [11]• Insertional mutagenesis (mainly LV) [3]• Difficult and expensive to manufacture at scale [13] | • Transient expression (for mRNA)• Off-target delivery can cause toxicity (e.g., hepatotoxicity) [16]• Requires optimization for tissue targeting [3] |
| Ideal Use Cases | • Gene therapies requiring permanent correction (e.g., genetic disorders) [3]• Ex vivo cell engineering (e.g., CAR-T) [12] | • mRNA vaccines & short-term therapies [3]• Treatments requiring repeated dosing [3]• Systemic delivery & CRISPR gene editing [3] [13] |
Advanced Experimental Protocols in LNP Research
Protocol: ASSET for Targeted mRNA Delivery
A major challenge for LNPs is precise tissue targeting. The Antibody-Specific Targeted LNP (ASSET) system was developed to overcome the suboptimal antibody orientation from conventional conjugation chemistries like EDC/NHS, which randomly attach via lysine residues and can inactivate the antigen recognition domain [17].
Methodology:
- Nanobody Engineering: An antibody-capturing nanobody (TP1107) is engineered. Using transmission electron microscopy (TEM), the precise binding site to the Fc domain of an IgG is determined.
- Site-Specific Modification: A synthetic amino acid (p-azido-phenylalanine, azPhe) is incorporated at the identified optimal site (Gln15) in TP1107, creating TP1107optimal. This is achieved using a genomically recoded E. coli host system [17].
- Lipid Conjugation: TP1107optimal is conjugated to DSPE-PEG2000-DBCO lipid via a click chemistry reaction between the DBCO group and the azide. This creates a lipid-PEG-nanobody construct.
- LNP Functionalization: The lipid-PEG-nanobody construct is incubated with pre-formed LNPs, inserting into the LNP membrane via its DSPE anchor.
- Antibody Capture: The functionalized LNPs are incubated with the desired targeting antibody, which is captured by the surface-exposed nanobody in an optimal orientation, ready for cell-specific delivery [17].
Data & Outcome: This method resulted in protein expression levels more than 1,000 times higher than non-targeted LNPs and more than 8 times higher than conventional antibody functionalization techniques, demonstrating highly efficient in vivo T cell targeting [17].
LNP Antibody Capture System Workflow
Protocol: Overcoming STING Pathway Activation for DNA-LNPs
A long-standing barrier to DNA-LNP therapy was lethal immune activation. Research discovered that standard DNA-LNPs trigger the STING (Stimulator of Interferon Genes) pathway, a defensive mechanism that causes severe inflammation [14].
Methodology:
- Problem Identification: Researchers established that loading DNA into standard mRNA-LNP formulations was lethal to 100% of healthy mice in lab tests, confirming a critical safety barrier.
- Mechanism Investigation: The inflammatory reaction was traced to the activation of the innate immune cGAS-STING pathway by the delivered DNA.
- Solution Development: Instead of modifying the DNA nucleotides (as done for mRNA), the team focused on the LNP composition. They incorporated a natural anti-inflammatory molecule, nitro-oleic acid (NOA), into the DNA-carrying particles.
- Validation: The safety and efficacy of the NOA-modified DNA-LNPs were tested in vivo. The modified particles completely eliminated the fatal reactions, with all test mice surviving [14].
Data & Outcome: This advancement enabled treated cells to produce the intended therapeutic proteins for about six months from a single dose, a significant duration compared to the short lifespan of mRNA therapies. It also allows for larger genetic payloads and more precise targeting compared to viral methods [14].
{width=760px} DNA-LNP Safety Breakthrough Pathway
The Scientist's Toolkit: Key Research Reagents
Successful research and development in gene delivery rely on a suite of critical reagents and technologies.
Table 3: Essential Research Reagents and Materials
| Reagent/Material | Function in Research | Specific Examples / Notes |
|---|---|---|
| Ionizable Lipids | Core component of LNPs; enables nucleic acid encapsulation and endosomal escape [17] [16]. | DLin-MC3-DMA (MC3), SM102 [17]. AI is used to design novel ionizable lipids with programmable pKa [16]. |
| Helper Lipids | Provide structural integrity to the LNP [17]. | DSPC, DOPE, Cholesterol [17]. |
| PEGylated Lipids | Enhance particle stability and circulation time; can influence targeting [17]. | DMG-PEG2000 (shorter C14 chain), DSPE-PEG2000 (longer C18 chain) [17]. PEG-lipids are also used for conjugating targeting ligands. |
| Targeting Ligands | Enable active targeting to specific cell types by binding to surface receptors. | Antibodies, nanobodies (e.g., TP1107 [17]), or small molecules (e.g., GalNAc for hepatocytes [12]). |
| Viral Vectors (AAV, LV) | Gold standard for high-efficiency gene delivery and long-term expression in gene therapy research. | AAV serotypes (e.g., AAV8, AAVrh74) for in vivo delivery; Lentivirus for ex vivo cell modification (e.g., CAR-T) [11] [12]. |
| AI/ML Platforms | Accelerate LNP design by predicting structure-property relationships and optimizing formulations virtually. | Graph Neural Networks (GNNs) for virtual screening; Generative Adversarial Networks (GANs) for de novo lipid design [16]. |
The field of gene delivery is rapidly evolving beyond a simple competition between viral and non-viral vectors. Future directions point toward hybrid approaches and intelligent design [3] [16].
- AI-Driven Optimization: Artificial intelligence is poised to revolutionize LNP development. Machine learning models can perform virtual screening of millions of lipid combinations, dramatically reducing development time from 6-12 months to a fraction of that, while also improving targeting specificity and reducing off-target effects [16].
- Combination Strategies: Researchers may leverage the strengths of both platforms—for example, using LNPs for initial or repeated dosing and viral vectors for sustained expression in specific tissues [3].
- Expanding Therapeutic Horizons: The advent of safe DNA-LNPs and advanced targeting technologies is set to broaden the application of gene therapies from rare diseases to common chronic conditions, such as heart disease and cancer [14].
In conclusion, both viral vectors and LNPs are powerful tools in the gene therapy arsenal. Viral vectors remain the established choice for therapies requiring long-term, permanent gene expression and have a robust track record of clinical success. LNPs, however, offer distinct advantages in safety, manufacturing scalability, and versatility, making them particularly suitable for vaccines, transient therapies, and treatments requiring re-dosing. The choice between them is not a matter of superiority but of strategic alignment with the therapeutic goal, target tissue, and desired duration of effect.
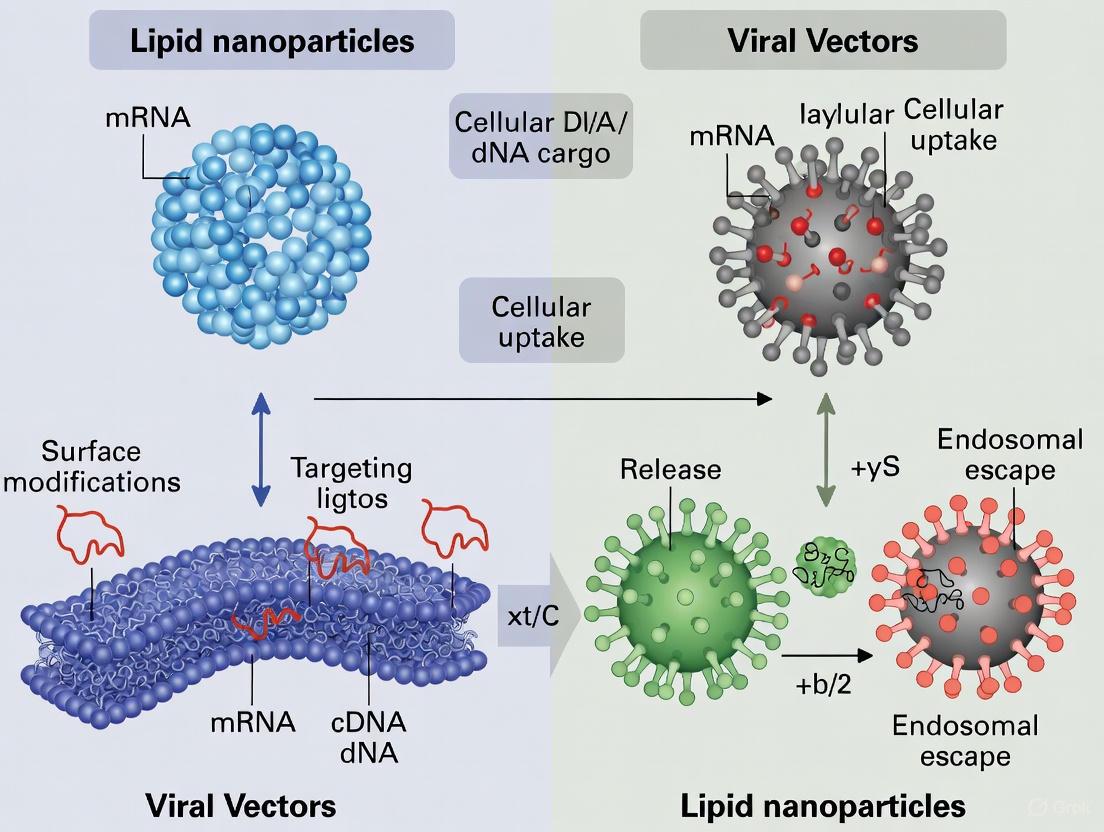
In the field of gene delivery, the efficacy and safety of a therapeutic are fundamentally dictated by its delivery vehicle. Lipid nanoparticles (LNPs) and viral capsids represent two dominant classes of delivery systems, each with a distinct structural blueprint that determines their function, capabilities, and limitations. LNPs are synthetic, spherical nanoparticles, while viral capsids are the protein shells of engineered viruses. Understanding their precise composition is not merely an academic exercise; it is crucial for researchers and drug development professionals to rationally select and optimize vectors for specific gene therapy applications. This guide provides a detailed, objective comparison of their structural compositions, supported by experimental data and characterization methodologies.
Core Structural Components and Their Functions
The fundamental architectures of LNPs and viral capsids arise from the assembly of their constituent parts, which directly correlate to their performance in gene delivery.
Composition of Lipid Nanoparticles (LNPs)
LNPs are sophisticated synthetic assemblies typically ranging from 50 to 150 nm in diameter [18] [19]. Their formulation is a precise mixture of four key lipidic components, each playing a critical role in stability, delivery, and release.
- Ionizable Lipids: This is the most crucial functional component. These lipids are positively charged at acidic pH during formulation, enabling efficient encapsulation of negatively charged nucleic acids, but are neutral at physiological pH, reducing toxicity [19] [20]. Their protonation in the acidic environment of the endosome is key to destabilizing the endosomal membrane and facilitating the release of the genetic payload into the cytoplasm. Examples include ALC-0315 and SM-102 [18] [19].
- Phospholipids: These lipids, such as DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), act as structural lipids that form the main bilayer structure of the nanoparticle, contributing to its stability and integrity [18] [20].
- Cholesterol: Cholesterol is a stability lipid that integrates into the LNP bilayer. It enhances structural integrity, improves packing efficiency, and facilitates membrane fusion, thereby aiding in cellular uptake and endosomal escape [18] [20].
- PEGylated Lipids: These lipids, like DMG-PEG2000, are located on the surface of the LNP. They play a dual role: they control particle size and prevent aggregation during storage and manufacturing, and they reduce nonspecific interactions in vivo by creating a hydrophilic layer [19] [20]. The PEG lipid is designed to dissociate after administration to allow cellular uptake.
The following diagram illustrates the assembly and structure of a typical LNP.
Composition of Viral Capsids
Viral vectors are engineered from viruses, with their capsids providing the natural machinery for efficient cellular entry. The specific protein composition varies by virus type, defining its tropism and behavior.
- Adeno-Associated Virus (AAV) Capsids: AAVs are small (~20 nm), non-enveloped viruses with an icosahedral protein capsid [1] [21]. The capsid is composed of three viral proteins (VP1, VP2, and VP3) that assemble in a specific ratio. These proteins determine the vector's tropism—its ability to infect specific tissues like liver, muscle, or neurons—and are a key target for engineering to alter targeting and evade pre-existing immune responses [21].
- Lentivirus (LV) Capsids: Lentiviruses are enveloped viruses, meaning their capsid is surrounded by a lipid bilayer derived from the host cell [1]. This envelope is studded with viral glycoproteins (e.g., VSV-G) that are essential for recognizing and entering target cells. The internal core contains the viral genome and enzymes. The ability of LVs to integrate into the host genome enables long-term gene expression but carries a risk of insertional mutagenesis [1] [21].
- Adenovirus (AdV) Capsids: Adenoviruses are larger, non-enveloped viruses with an icosahedral capsid. Their surface is characterized by fiber proteins that protrude from the vertices, which are critical for binding to host cell receptors. AdV vectors offer high transgene expression and large cargo capacity but often trigger strong immune responses [1] [21].
The schematic below generalizes the structure of an enveloped viral vector like Lentivirus.
Comparative Structural Analysis
The distinct compositions of LNPs and viral capsids translate into direct differences in their physical characteristics, payload capacity, and functional outcomes. The table below summarizes a direct, quantitative comparison based on the available experimental data.
Table 1: Direct Comparison of LNP and Viral Capsid Attributes
| Feature | Lipid Nanoparticles (LNPs) | Viral Vectors (e.g., AAV, Lentivirus) |
|---|---|---|
| Primary Components | Synthetic ionizable lipids, phospholipids, cholesterol, PEG-lipids [19] [20] | Viral proteins (e.g., VP1-3 for AAV), sometimes a host-derived lipid envelope [1] |
| Typical Size Range | 50–150 nm [18] [19] | ~20–100 nm (varies by virus; AAV ~20 nm) [21] |
| Payload Type | mRNA, siRNA, saRNA, CRISPR-Cas9 RNA components [3] [19] | Primarily DNA; some RNA (e.g., Lentivirus) [1] |
| Payload Capacity | High. Can deliver large RNA payloads (>4.5 kb), including multiple RNAs for CRISPR [19]. | Limited. AAV cargo capacity is ~4.7 kb, restricting delivery of large genes [21]. |
| Immunogenicity | Lower. Suitable for repeat dosing, though anti-PEG immunity can occur [3] [22]. | Higher. Pre-existing and treatment-induced immunity can limit efficacy and re-dosing [3] [21]. |
| Expression Duration | Transient. Ideal for short-term expression (e.g., vaccines, transient editing) [3]. | Long-term/Stable. Can provide sustained expression; integrating vectors (LV) can be permanent [3] [1]. |
| Manufacturing | Highly scalable and rapid. Microfluidic mixing allows production in days [3] [19]. | Complex and time-consuming. Requires cell culture, takes several weeks, difficult to scale [21] [19]. |
Experimental Characterization of Composition and Structure
Rigorous characterization is essential to confirm the structure, stability, and functionality of these gene delivery vectors. The following experimental protocols are standard in the field.
Key Methodologies for LNP Analysis
Protocol 1: Assessing LNP Size and Stability Using Dynamic Light Scattering (DLS)
- Objective: To determine the average particle size, polydispersity index (PDI, a measure of size distribution), and physical stability of LNPs under storage conditions [18].
- Materials: LNP formulation, dispersant (e.g., 8.7% sucrose in Tris buffer, pH 7.4), DLS instrument (e.g., Malvern Nano ZS), polystyrene cuvettes [18].
- Procedure:
- Dilute the LNP sample in an appropriate dispersant (e.g., 50 µL LNPs into 1450 µL dispersant) to achieve optimal scattering intensity.
- Transfer the diluted sample to a polystyrene cuvette.
- Place the cuvette in the DLS instrument pre-equilibrated to 25°C.
- Set parameters: dispersant refractive index, viscosity, equilibration time of 120 s.
- Perform measurement. The instrument analyzes fluctuations in scattered light to calculate hydrodynamic diameter and PDI.
- Data Interpretation: A lower PDI (<0.2) indicates a more monodisperse, homogeneous sample. Studies show that LNPs in the 80–100 nm range often demonstrate superior stability during long-term storage at 4°C and -20°C [18].
Protocol 2: Visualizing LNP Morphology via Cryo-Electron Microscopy (Cryo-TEM)
- Objective: To observe the internal and external structure, lamellarity, and morphology of LNPs in a frozen-hydrated, near-native state [18].
- Materials: LNP formulation, cryo-TEM instrument, holey carbon grid, vitrification apparatus (plunger).
- Procedure:
- Apply a small volume (3-5 µL) of the LNP sample onto a glow-discharged holey carbon grid.
- Blot away excess liquid with filter paper to form a thin liquid film across the grid holes.
- Rapidly plunge-freeze the grid into a cryogen (typically liquid ethane) to vitrify the water, preventing ice crystal formation.
- Transfer the grid under liquid nitrogen to the cryo-TEM holder and insert into the microscope.
- Image the particles at low dose and under cryo-conditions to minimize radiation damage.
- Data Interpretation: Cryo-TEM micrographs reveal the spherical structure of LNPs, the integrity of the lipid bilayer, and the presence of an electron-dense core, confirming successful mRNA encapsulation [18].
Key Methodologies for Viral Vector Analysis
Protocol 3: Determining Viral Titer and Purity
- Objective: To quantify the concentration of functional viral vector particles and assess purity relative to non-infectious or empty capsids.
- Materials: Viral vector prep, qPCR kit, cell line permissive to the virus, tissue culture reagents, SDS-PAGE equipment.
- Procedure (Functional Titer):
- Transduction: Serially dilute the viral vector and apply to permissive cells.
- Analysis: After an appropriate period, analyze cells for transgene expression (e.g., by flow cytometry if it's a fluorescent protein) or resistance to selection antibiotics.
- Calculation: The functional titer (e.g., Transducing Units/mL, TU/mL) is calculated based on the dilution and percentage of positive cells.
- Procedure (Physical Titer - qPCR):
- Digestion: Treat the viral prep with DNase to degrade any unpackaged DNA.
- Lysis: Lyse the virus particles to release the encapsulated genome.
- qPCR: Perform qPCR using primers specific to a conserved region of the viral genome (e.g., the WPRE element in LV). Compare to a standard curve of known concentration.
- Data Interpretation: The ratio of physical titer (genome copies/mL) to functional titer (TU/mL) indicates the fraction of particles that are functional. A high ratio suggests many empty or defective particles.
The Scientist's Toolkit: Essential Research Reagents
Successful research and development in gene delivery rely on a suite of critical reagents and materials. The following table details key solutions used in the featured experiments and general formulation workflows.
Table 2: Essential Research Reagents for Vector Development
| Reagent / Material | Function and Application |
|---|---|
| Ionizable Lipids (e.g., SM-102, ALC-0315) | The core functional lipid for mRNA encapsulation and endosomal escape in LNP formulations [18] [19]. |
| PEG-Lipids (e.g., DMG-PEG2000) | Used to control LNP size, improve colloidal stability, and reduce nonspecific protein adsorption during storage and in vivo administration [19] [20]. |
| Structural Lipids (DSPC, Cholesterol) | Provide the structural backbone and bilayer stability for LNPs, mimicking biological membranes and aiding in fusion [18] [20]. |
| Microfluidic Mixers | Essential equipment for the scalable and reproducible production of LNPs by rapidly mixing lipid and aqueous phases in a controlled manner [20]. |
| Cryo-TEM and DLS Instrumentation | Critical analytical tools for characterizing the morphology, size, and size distribution of both LNPs and viral particles [18]. |
| Quant-it RiboGreen RNA Assay Kit | A fluorescent assay used to accurately determine RNA encapsulation efficiency within LNPs by measuring free vs. total RNA [22]. |
| Viral Envelope Proteins (e.g., VSV-G) | Commonly used pseudotyping glycoproteins for Lentiviral vectors, which determine tropism and enable efficient transduction of a broad range of cell types [1]. |
The choice between LNPs and viral capsids is not a matter of superiority, but of strategic alignment with therapeutic goals. LNP's synthetic, modular composition offers flexibility, scalability, and a favorable safety profile for transient expression, making it ideal for vaccines and in vivo gene editing. In contrast, the complex, evolved biology of viral capsids provides unrivalled delivery efficiency and potential for long-term gene expression, which is necessary for many monogenic disorders, albeit with challenges related to immunogenicity and manufacturing. Future directions point toward hybrid and engineered approaches—such as cell-specific targeting ligands for LNPs and engineered capsids to evade immunity—that leverage the strengths of both blueprints to unlock the full potential of genetic medicine.
Introducing foreign genetic material into cells is a cornerstone of modern biological research and therapeutic development. The two predominant methods for achieving this are viral transduction and non-viral transfection, specifically using Lipid Nanoparticles (LNPs). While both share the same ultimate goal, their fundamental mechanisms of action—from cellular entry to intracellular trafficking and payload delivery—are profoundly different. Viral transduction leverages billions of years of viral evolution to achieve highly efficient, receptor-mediated entry and, in some cases, genomic integration. [23] [24] In contrast, LNP-mediated transfection utilizes synthetic lipid-based carriers to encapsulate and protect genetic payloads, facilitating delivery through endocytic pathways and subsequent endosomal escape. [25] [3] This guide provides a detailed, objective comparison of these mechanisms, supported by experimental data and protocols, to inform researchers and drug development professionals in selecting the optimal system for their specific applications.
Core Mechanisms of Action
Viral Vector Transduction
Transduction is a process that utilizes viral vectors to deliver genetic material into a cell. [23] Engineered viral vectors are modified to carry a therapeutic transgene while typically being rendered replication-incompetent. The process is characterized by its high efficiency and reliance on specific receptor-ligand interactions.
Figure 1: Viral Transduction Pathways for AAV and Lentivirus Vectors. The process begins with receptor binding and culminates in different outcomes based on the viral serotype.
The key steps in viral transduction, as illustrated in Figure 1, involve:
- Receptor Binding: Viral vectors bind to specific cell surface receptors (e.g., AAV2 uses heparan sulfate proteoglycan), which determines their tropism and tissue specificity. [24]
- Cellular Uptake: The vector-receptor complex enters the cell via receptor-mediated endocytosis or, in some cases, direct membrane fusion. [24]
- Uncoating: The viral capsid is degraded within the endosome, releasing the viral genome into the cytoplasm. [26]
- Genome Processing and Trafficking: The genetic payload is processed and trafficked to the nucleus. For lentiviruses (RNA viruses), this involves reverse transcription and nuclear import of the resulting DNA. For AAV (ssDNA virus), this involves second-strand synthesis of its single-stranded DNA genome. [24] [26]
- Transgene Fate: Lentiviral vectors integrate their DNA into the host cell genome, leading to stable, long-term transgene expression. AAV vectors typically persist as non-integrating episomes in the nucleus, providing durable expression in non-dividing cells. [24] [26]
LNP-Mediated Transfection
Transfection is the process of introducing nucleic acids into cells using non-viral methods, with Lipid Nanoparticles (LNPs) being a leading technology. [23] [27] LNPs are synthetic, multi-component carriers that encapsulate and protect genetic material. Their mechanism is distinct from viral vectors and does not involve specific receptor targeting in its basic form.
Figure 2: LNP-Mediated Transfection Pathway and Intracellular Barriers. The process is less efficient than viral transduction due to several intracellular barriers.
The key steps in LNP-mediated transfection, as illustrated in Figure 2, involve:
- Cell Membrane Interaction: Positively charged LNPs interact with the negatively charged cell membrane, leading to adsorption and initiation of uptake. [13]
- Cellular Uptake: LNPs are internalized via endocytosis, primarily through clathrin-mediated or other endocytic pathways, forming an endosome. [13]
- Endosomal Trafficking and Acidification: The endosome matures and its internal pH drops. This acidic environment (pH ~6.0-6.5) protonates the ionizable lipids within the LNP, giving them a positive charge. [28]
- Endosomal Escape: The protonated ionizable lipids promote fusion with or destabilization of the endosomal membrane. This is often visualized by the recruitment of galectin proteins, which mark damaged endosomal membranes. [28] Recent studies show that only a small fraction of RNA is actually released from these galectin-marked endosomes, representing a major efficiency barrier. [28]
- Payload Release: The nucleic acid (e.g., mRNA, siRNA) is released into the cytosol, where it can be translated by ribosomes (mRNA) or engage the RNA-induced silencing complex (siRNA). The LNP components and payload can segregate during endosomal sorting, further reducing efficiency. [28]
Comparative Experimental Data
To objectively compare the performance of viral vectors and LNPs, key quantitative data from the literature is summarized in the tables below.
Table 1: Comparison of Key Characteristics Between Viral Vectors and LNPs
| Characteristic | Viral Vectors (LV, AAV) | Lipid Nanoparticles (LNPs) | Experimental Context & Notes |
|---|---|---|---|
| Fundamental Mechanism | Receptor-mediated transduction [24] | Chemical/physical transfection [23] [27] | Defined by the presence or absence of a viral capsid. |
| Primary Uptake Route | Receptor-mediated endocytosis / Membrane fusion [24] | Endocytosis (e.g., clathrin-mediated) [13] | Viral entry is highly specific; LNP entry is generally non-specific. |
| Payload Location | Nuclear (for DNA vectors) [24] | Cytosolic [3] | Suits different applications: gene editing/stable expression (viral) vs. mRNA/siRNA (LNP). |
| Expression Kinetics | Long-term (stable integration or episomal persistence) [23] [24] | Transient (hours to days) [3] | LNP kinetics ideal for vaccines; viral for correcting genetic defects. |
| Typical Payload | DNA (ssDNA for AAV, RNA for LV) [24] | RNA (mRNA, siRNA) or DNA [25] [3] | LNPs have more versatile payload capacity. |
| Immunogenicity | Moderate to High (can trigger immune responses) [23] [3] | Low to Moderate (lower immunogenicity) [3] [13] | LNP's lower immunogenicity allows for repeated dosing. [3] |
Table 2: Quantitative Performance and Practical Considerations
| Parameter | Viral Vectors (LV, AAV) | Lipid Nanoparticles (LNPs) | References & Data Source |
|---|---|---|---|
| Delivery Efficiency | High (evolved infection mechanism) [3] | Variable, can be high but faces intracellular barriers [28] [3] | A key inefficiency for LNPs is limited endosomal escape. [28] |
| Tropism / Targeting | High (natural and engineered tropism) [24] | Lower, but can be engineered with ligands [3] [13] | Viral serotypes (e.g., AAV2, AAV9) have innate tissue preferences. [24] |
| Cargo Capacity | Limited (~4.7 kb for AAV) [23] | Large (>10 kb), more versatile [25] [3] | AAV's small capacity is a major limitation, overcome by dual-vector approaches. [9] |
| Manufacturing | Complex, time-consuming, costly [3] [13] | Simplified, scalable, commercially viable [3] [13] | LNP scalability was demonstrated during COVID-19 vaccine production. [3] |
| Endosomal Escape Efficiency | Highly efficient (viral uncoating) [24] | Inefficient (rate-limiting step); only a fraction of RNA is released [28] | Live-cell microscopy showed only ~20% of mRNA-LNP damaged endosomes contained detectable mRNA. [28] |
| Safety Profile | Risk of insertional mutagenesis (LV) and immunogenicity [23] [3] | Safer; no genomic integration, lower immunogenicity [25] [3] | LNP toxicity is primarily associated with lipid composition. [3] |
Detailed Experimental Protocols
To ensure reproducibility and provide a deeper understanding of the data generating these comparisons, key experimental methodologies are outlined below.
Protocol for Quantifying LNP Endosomal Escape Efficiency
This protocol is based on live-cell and super-resolution microscopy studies that directly visualize the inefficiencies in LNP-mediated cytosolic delivery. [28]
Objective: To quantify the efficiency of endosomal escape and RNA release for MC3-based LNPs. Key Reagents:
- Fluorescently labeled siRNA or mRNA (e.g., AlexaFluor 647-siRNA, Cy5-mRNA).
- MC3-based LNPs formulated with the labeled RNA.
- Cell line of interest (e.g., HeLa, HEK-293).
- Galectin-9 fluorescent protein marker (e.g., Galectin-9-GFP) to detect endosomal membrane damage.
- Live-cell imaging medium.
Methodology:
- Cell Preparation: Seed cells onto glass-bottom imaging dishes and culture until they reach 60-80% confluency.
- Transfection: Treat cells with a defined dose of fluorescent RNA-LNPs (e.g., 50 nM for siRNA-LNPs, 0.75 µg/mL for mRNA-LNPs) in live-cell imaging medium. [28]
- Live-Cell Imaging: Use fast live-cell microscopy to image cells over time (starting from 1 hour post-transfection). Capture simultaneous channels for the fluorescent RNA signal (e.g., Cy5) and the galectin-9 damage sensor (e.g., GFP).
- Image Analysis:
- Identify individual endosomes that show de novo recruitment of galectin-9.
- For each galectin-9-positive endosome, quantify the presence or absence of the fluorescent RNA signal.
- Calculate the "hit rate" as the percentage of galectin-9-positive endosomes that contain a detectable RNA signal.
- Expected Outcome: The hit rate for siRNA-LNPs is typically 67-74%, while for mRNA-LNPs it is significantly lower, around 20%, indicating a major barrier in productive mRNA cargo release. [28]
Protocol for Evaluating Viral Transduction Efficiency and Specificity
Objective: To determine the transduction efficiency and tropism of a specific viral vector (e.g., AAV or Lentivirus). Key Reagents:
- Recombinant viral vector (e.g., AAV2, AAV9, LV) carrying a reporter gene (e.g., GFP, luciferase).
- Target cell lines (including primary cells if applicable) with known receptor expression profiles.
- Appropriate cell culture media and supplements.
- Transduction enhancers (e.g., polybrene for LV).
- Flow cytometer or fluorescence microscope for analysis.
Methodology:
- Cell Preparation: Seed different target cell lines at an optimal density.
- Viral Transduction: Treat cells with a range of viral vector doses (Multiplicity of Infection - MOI) in the presence or absence of specific transduction enhancers. Include controls (non-transduced cells).
- Incubation and Expression: Incubate cells for the required time to allow for transgene expression (e.g., 48-72 hours).
- Efficiency Analysis:
- For reporter genes: Analyze cells via flow cytometry to quantify the percentage of GFP-positive cells (transduction efficiency) and mean fluorescence intensity (expression level).
- For functional genes: Use RT-qPCR to measure transgene mRNA levels or a functional assay relevant to the delivered gene.
- Specificity/Tropism Confirmation: Perform blocking experiments with excess soluble receptor (if available) to confirm receptor-specific entry. Compare efficiency across cell lines with varying receptor expression levels.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Resources for Gene Delivery Research
| Reagent / Resource | Function / Description | Example Applications |
|---|---|---|
| Adeno-associated Virus (AAV) Vectors | Small, single-stranded DNA vectors with low immunogenicity and multiple serotypes for specific tropism (e.g., AAV2 for liver, AAV9 for CNS). [24] | In vivo gene therapy for retinal diseases (Luxturna), hereditary hearing loss. [9] |
| Lentiviral (LV) Vectors | RNA vectors that integrate into the host genome, enabling stable, long-term transgene expression in dividing and non-dividing cells. [24] | Generation of stable cell lines, CAR-T cell therapy ex vivo. [9] [24] |
| Ionizable Cationic Lipids (e.g., MC3) | Key component of LNPs; protonated in acidic endosomes to disrupt the endosomal membrane and facilitate cargo escape. [28] | Formulating LNPs for siRNA (Patisiran) and mRNA therapeutics. [28] |
| Galectin-9 Fluorescent Marker | A sensitive biosensor that recruits to sites of endosomal membrane damage, used to visualize and quantify LNP-induced endosomal escape events. [28] | Live-cell imaging assays to study the efficiency and mechanisms of endosomal escape. [28] |
| Polyethyleneimine (PEI) | A cationic polymer that condenses nucleic acids into "polyplexes" for delivery; can be used for both DNA and RNA transfection. [25] [24] | A common, high-efficiency (but sometimes cytotoxic) transfectant for in vitro studies in easy-to-transfect cell lines. [25] |
| Electroporation Systems | Physical method using electrical pulses to create transient pores in the cell membrane for nucleic acid entry. [25] [24] | Transfection of hard-to-transfect cells like primary cells and stem cells. [25] [24] |
The fundamental actions of viral vector transduction and LNP-mediated transfection are distinct, leading to clear trade-offs that dictate their application in research and therapy. Viral vectors excel in delivering high-efficiency, long-term, and targeted gene expression, making them indispensable for in vivo gene therapy and creating stable cell models. Their limitations include immunogenicity, limited cargo capacity, and complex manufacturing. [23] [3] [24] LNPs offer a safer, more versatile, and scalable platform ideal for transient expression applications, such as mRNA vaccines and siRNA silencing. Their primary challenge is overcoming intracellular barriers to achieve consistently high delivery efficiency across diverse cell types. [25] [28] [3] The choice between these systems is not a matter of superiority but of strategic alignment with the experimental or therapeutic goal. Future progress will likely involve hybrid approaches and continued innovation to overcome the inherent limitations of each platform, further expanding the toolbox for genetic medicine.
Clinical Translation and Therapeutic Applications in Practice
The choice between in vivo and ex vivo therapeutic strategies is a fundamental consideration in advanced therapy medicinal product (ATMP) development. This decision is intrinsically linked to the selection of a gene delivery platform, each with distinct implications for research, manufacturing, and clinical application. Lipid nanoparticles (LNPs) and viral vectors represent two leading technologies that enable these paradigms, offering contrasting profiles in efficiency, safety, and therapeutic durability. This guide provides an objective comparison of these platforms, mapping their performance characteristics to appropriate treatment strategies across diverse disease contexts to inform research and drug development decision-making.
The table below synthesizes the core relationship between delivery platforms and therapeutic paradigms, highlighting how inherent platform characteristics direct their application toward specific treatment strategies.
Table 1: Mapping Delivery Platforms to Therapeutic Paradigms
| Feature | In Vivo Paradigm | Ex Vivo Paradigm |
|---|---|---|
| Primary Platform | Lipid Nanoparticles (LNPs) [29] [3] | Viral Vectors (Lentivirus, AAV) & Electroporation [30] [29] |
| Description | Therapy administered directly to the patient; editing occurs inside the body [29] | Patient cells harvested, modified outside the body, then re-infused [29] |
| Therapeutic Duration | Transient expression (e.g., mRNA delivery) [3] | Long-term or permanent expression (e.g., genomic integration) [3] |
| Key Advantages | Non-invasive administration; suitable for inaccessible tissues; scalable production [29] [3] | High precision editing; controlled conditions; reduced immune concerns [29] |
| Key Limitations | Potential off-target effects; lower efficiency in some tissues; immune reactions [30] | Complex, costly manufacturing; limited to cell types that can be cultured [29] |
| Therapeutic Examples | NTLA-2001 (TTR Amyloidosis), CTX310 (Cardiovascular Disease) [29] | Casgevy (Sickle Cell Disease), Lyfgenia (Sickle Cell Disease) [29] |
Technical Performance and Experimental Data
A direct comparison of technical performance metrics is essential for platform selection. The following table summarizes quantitative and qualitative data on the key characteristics of LNPs and viral vectors.
Table 2: Technical Performance Comparison of Lipid Nanoparticles vs. Viral Vectors
| Performance Metric | Lipid Nanoparticles (LNPs) | Viral Vectors (AAV, Lentivirus) |
|---|---|---|
| Delivery Efficiency | High for liver targets; improving for extrahepatic tissues [31] [32] | Very high, particularly with AAV and lentiviral vectors [3] |
| Payload Capacity | Versatile; suitable for mRNA, siRNA, CRISPR components [3] | Limited cargo size, especially for AAV [29] [3] |
| Immune Response | Lower immunogenicity; suitable for repeated dosing [3] | Higher immunogenicity; risk of pre-existing immunity [3] |
| Tissue Targeting | Improving with novel formulations & membrane modifications [31] [33] | Naturally high tropism; can be engineered for specificity [34] [33] |
| Gene Integration | Typically non-integrating; transient expression [3] | Lentiviruses integrate; AAV mostly episomal (long-term expression) [3] |
| Manufacturing Scalability | Highly scalable; demonstrated at global scale [29] [3] | Complex and costly to scale; challenges in consistency [3] |
| Key Safety Concerns | Reactogenicity, lipid toxicity [35] | Insertional mutagenesis, immune toxicity [3] |
| Representative Therapy | mRNA COVID-19 Vaccines, NTLA-2001 [35] [29] | Zolgensma, Luxturna, Lyfgenia [29] |
Experimental Protocols and Workflows
Ex Vivo Workflow: CRISPR-Edited Cell Therapies
The ex vivo paradigm, used in FDA-approved therapies like Casgevy, involves a multi-stage process where genetic modification occurs outside the patient's body [29].
Key Methodological Details:
- Cell Harvest: CD34+ hematopoietic stem and progenitor cells (HSPCs) are collected from the patient via apheresis following mobilization, or directly from bone marrow [29].
- Ex Vivo Gene Editing: Cells are transfected using electroporation (for CRISPR ribonucleoproteins) or transduced with lentiviral vectors. For Casgevy, electroporation delivers the CRISPR-Cas9 system to precisely edit the BCL11A gene enhancer in HSPCs [29].
- Cell Expansion and Quality Control: Edited cells are cultured ex vivo to expand the population and undergo rigorous testing to ensure viability, purity, and editing efficiency before reinfusion.
- Patient Conditioning and Reinfusion: Patients receive myeloablative conditioning (e.g., busulfan) to clear marrow space. The edited HSPCs are then infused back into the patient to engraft and reconstitute the hematopoietic system with genetically corrected cells [29].
In Vivo Workflow: Systemic LNP Administration
The in vivo paradigm, exemplified by therapies like NTLA-2001, delivers the genetic medicine directly to the patient via systemic administration [29].
Key Methodological Details:
- LNP Formulation: LNPs are synthesized to encapsulate mRNA encoding CRISPR-Cas9 and a single guide RNA (sgRNA) targeting the therapeutic gene (e.g., the TTR gene for NTLA-2001). Microfluidic mixing is a standard method for producing monodisperse, reproducible LNPs [32] [29].
- Systemic Administration and Targeting: The formulated LNPs are administered intravenously. Following IV injection, many current LNP systems naturally accumulate in the liver via apolipoprotein E (ApoE)-mediated uptake, effectively targeting hepatocytes [31].
- Intracellular Delivery and Endosomal Escape: After cellular uptake via endocytosis, the ionizable lipids within the LNPs become protonated in the acidic endosomal environment. This promotes a hexagonal lipid phase structure that disrupts the endosomal membrane, releasing the genetic payload into the cytoplasm [32].
- Gene Editing and Phenotypic Effect: The delivered Cas9 mRNA is translated into functional protein, which complexes with the sgRNA to form the active nuclease. This complex introduces a double-strand break in the target genomic DNA, leading to gene knockout via non-homologous end joining (NHEJ). Successful knockout of the TTR gene in hepatocytes results in a sustained reduction of pathogenic TTR protein serum levels, as demonstrated in clinical trials for NTLA-2001 [29].
Advanced Engineering and Screening Methodologies
High-Throughput LNP Development
The traditional, sequential approach to LNP formulation is being transformed by integrated high-throughput strategies. These approaches leverage automation and advanced screening to rapidly identify optimal candidates from vast combinatorial libraries [32].
Key Components of the Workflow:
- Combinatorial Lipid Libraries: Automated synthesis enables the rapid generation of hundreds to thousands of unique ionizable lipids with systematic structural variations, creating a vast chemical space for exploration [32].
- Automated Microfluidic Formulation: Microfluidic chips enable the parallel synthesis of LNP libraries in multi-well plates (up to 384 per plate), ensuring monodispersity and high batch-to-batch consistency with minimal reagent consumption [32].
- High-Throughput Characterization (HTC): Automated systems using multi-well dynamic light scattering (DLS), spectroscopy, and other techniques rapidly profile LNP size, stability, charge, and encapsulation efficiency across thousands of formulations [32].
- High-Throughput Screening (HTS): Multiplexed in vitro assays and in vivo barcoding strategies assess cellular uptake, transfection efficiency, cytotoxicity, and biodistribution to identify candidates with desirable biological performance [32].
- Machine Learning Integration: Data from HTC and HTS feed into predictive models like COMET (Composite Material Transformer), a transformer-based neural network trained on large LNP datasets. COMET can predict LNP efficacy and guide the design of improved formulations in an iterative, closed-loop process [36].
Directed Evolution of Viral Vectors
To overcome limitations such as off-target transduction and immune recognition, viral vectors can be engineered for enhanced specificity and efficiency through directed evolution [33].
Protocol for AAV Capsid Engineering via In Vivo Selection:
- Library Generation: Create a diverse library of AAV capsid mutants, for example, by inserting random 7-amino acid peptides into the capsid protein of a parental serotype like AAV9 [33].
- In Vivo Selection: Administer the mutant library systemically to animal models (e.g., transgenic Cre mice). Employ focused ultrasound blood-brain barrier opening (FUS-BBBO) in specific brain regions to enable localized viral entry [33].
- Recovery and Analysis: Recover viral DNA specifically from the targeted brain regions. Use Cre-dependent PCR to selectively amplify genomes that have successfully transduced neurons. Sequence the recovered capsid variants to identify enriched mutants [33].
- Validation: Package selected mutant capsids and validate their performance against the parental serotype. The goal is to identify variants with enhanced targeting specificity, reduced off-target organ transduction, and improved neuronal tropism [33].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of these paradigms requires a suite of specialized tools and reagents. The following table details key solutions for developing and optimizing gene delivery systems.
Table 3: Essential Research Reagents and Solutions for Gene Delivery Research
| Tool Category | Specific Examples | Function and Application |
|---|---|---|
| LNP Components | Ionizable Lipids (e.g., C12-200, LP01), PEG-lipids, Cholesterol, Helper Lipids (DOPE, DSPC) [32] [29] | Structural components of LNPs; ionizable lipids are critical for endosomal escape and efficacy [32]. |
| Microfluidic Systems | PreciGenome NanoGenerator, other commercial microfluidic chips [29] | Enable reproducible, high-throughput synthesis of monodisperse LNPs for research and pre-clinical testing [32] [29]. |
| Characterization Instruments | High-Throughput Dynamic Light Scattering (DLS), Microplate Readers, SAXS instruments [32] | Automated, multi-parametric physicochemical characterization of LNP libraries (size, PDI, stability, encapsulation) [32]. |
| Viral Vector Engineering Tools | AAV Capsid Libraries (e.g., 7-mer insertion libraries), Cre-transgenic Mice, Packaging Cell Lines [33] | Key reagents for directed evolution of viral vectors to achieve enhanced tissue tropism and reduced immunogenicity [33]. |
| Analytical & Screening Tools | Next-Generation Sequencing (NGS), Barcoded Library Strategies, In Vivo Imaging Systems (IVIS) [32] [33] | Critical for deconvoluting complex screening results, tracking biodistribution, and quantifying editing efficiency. |
| Cell Culture & Editing Reagents | Hematopoietic Stem Cell Media, CRISPR-Cas9 RNP Complexes, Electroporation Systems [29] | Essential for ex vivo gene editing workflows, enabling the modification and expansion of patient-derived cells [29]. |
The strategic selection between in vivo and ex vivo paradigms, and their corresponding delivery platforms, is guided by the specific therapeutic objective. The ex vivo approach, largely enabled by electroporation and viral vectors, offers a controlled, precise method for modifying cells outside the body and is the foundation of the first approved CRISPR therapies. In contrast, the in vivo paradigm, significantly advanced by LNP technology, provides a less invasive, directly administered, and highly scalable option, as demonstrated by therapies in development for liver and metabolic diseases.
Emerging technologies such as high-throughput screening, machine learning-guided LNP design [36], and directed evolution of viral capsids [33] are rapidly enhancing the performance of both platforms. The future of gene therapy will likely see a more refined application of each platform based on disease pathology and target tissue, and potentially even hybrid strategies that combine their respective strengths to achieve optimal therapeutic outcomes.
The debate between viral vector and non-viral delivery systems represents a central theme in gene therapy research. While lipid nanoparticles (LNPs) have gained prominence for RNA-based vaccines and therapeutics, viral vectors, particularly recombinant adeno-associated viruses (rAAVs), have established a proven track record with multiple FDA-approved treatments for rare diseases. These successful clinical applications highlight the unique advantages of viral vectors, including their ability to provide long-term transgene expression and their high tissue specificity, which are critical for addressing the root cause of many genetic disorders.
This guide objectively examines the performance of approved rAAV therapies, using Zolgensma and Luxturna as primary case studies, and compares them with emerging alternatives like LNPs, supported by experimental and clinical data.
The following table summarizes key FDA-approved rAAV-based gene therapies, their indications, and mechanisms of action.
| Therapy Name | Indication | Target Gene / Mechanism | Vector Serotype | Key Clinical Outcome |
|---|---|---|---|---|
| Zolgensma [37] | Spinal muscular atrophy (SMA) in pediatric patients <2 years | Survival motor neuron 1 (SMN1) gene replacement | AAV9 | Improves motor function and survival; sustained SMN protein expression |
| Luxturna [37] [38] | RPE65 mutation-associated retinal dystrophy | Retinal pigment epithelium-specific 65 kDa protein (RPE65) gene replacement | AAV2 | Improved functional vision; ~70% of patients maintained gains at 4 years [38] |
| Hemgenix [37] | Hemophilia B | Factor IX (FIX) gene replacement | AAV5 | Sustained therapeutic levels of FIX, reducing or eliminating need for prophylaxis |
| Elevidys [37] | Duchenne muscular dystrophy (DMD) in ambulatory children | Microdystrophin gene expression | AAVrh74 | Approved under accelerated approval; increased microdystrophin expression |
| Roctavian [37] | Severe hemophilia A | Factor VIII (FVIII) gene replacement | AAV5 | Sustained FVIII expression, reducing bleeding episodes |
| Kebilidi [37] | Aromatic L-amino acid decarboxylase (AADC) deficiency | AADC gene replacement | AAV2 | Improved motor and cognitive function |
Performance Analysis: Viral Vectors vs. Emerging Alternatives
Therapeutic Efficacy and Durability
rAAV vectors excel in providing long-lasting therapeutic effects due to their episomal persistence in non-dividing cells, leading to sustained transgene expression. For example, Luxturna has demonstrated durable functional vision improvements, with approximately 70% of patients maintaining gains up to four years post-treatment [38].
In vivo genome editing therapies using rAAV vectors also show promise for durable outcomes. The rAAV-based CRISPR system used in EDIT-101, an investigational therapy for Leber Congenital Amaurosis, was designed to create a permanent genomic correction [39].
Safety and Immunogenicity Profile
While rAAV therapies are generally safe, post-marketing surveillance has identified specific safety profiles that require careful management.
- Luxturna: Clinical trials showed largely mild to moderate adverse events (e.g., conjunctival hyperemia, cataract). However, post-marketing data revealed chorioretinal atrophy (CRA) in 13–50% of treated eyes, particularly in younger patients and often near the injection site [38].
- Common Challenges: Immune responses to the viral capsid or transgene can limit re-administration and, in some cases, impact efficacy and safety. Pre-existing immunity to certain AAV serotypes in human populations can also exclude eligible patients [39] [40].
Manufacturing and Scalability
Manufacturing complexity presents a significant challenge for viral vectors. The production of rAAVs involves complex processes using mammalian cell culture systems, which can be difficult to scale up for commercial supply [40]. Industry partnerships are increasingly requiring higher levels of Good Manufacturing Practice (GMP) compliance early in clinical development, adding to the complexity [40].
Experimental Insights: Direct Comparison of Delivery Platforms
The table below compares key technical and performance characteristics of viral and non-viral gene delivery platforms, based on data from preclinical and clinical studies.
| Parameter | rAAV Vectors | Lipid Nanoparticles (LNPs) | Electroporation |
|---|---|---|---|
| Packaging Capacity | Limited (<4.7 kb) [39] | Higher capacity for RNA and CRISPR ribonucleoproteins [41] | Wide compatibility [41] |
| Expression Durability | Long-term (episomal persistence) [39] | Transient (ideal for short-term editing) [41] [42] | Can be stable if genome integration occurs |
| Typical Administration | In vivo [37] [39] [38] | In vivo or ex vivo [41] [42] | Ex vivo only [41] |
| Primary Safety Concerns | Immunogenicity, off-target editing (CRISPR) [39] [38] | Transient inflammatory responses [41] | Low cell viability, restricted to ex vivo use [41] |
| Manufacturing Scalability | Complex and costly [40] | Highly scalable (e.g., microfluidics) [41] [43] | Not applicable for in vivo use |
| Clinical Proof of Concept | Multiple FDA-approved in vivo therapies [37] | FDA-approved vaccines; in vivo CRISPR therapies in trials (e.g., NTLA-2001) [41] | FDA-approved ex vivo therapy (Casgevy) [41] |
Detailed Experimental Protocols for Viral Vector Therapies
Protocol: Subretinal Administration of rAAV (Exemplified by Luxturna)
The subretinal injection technique is critical for the success of retinal gene therapies like Luxturna [38].
Key Methodological Details:
- Dosage: Luxturna is administered at a standardized dose of 1.5 × 10^11 vector genomes (vg) per eye [38].
- Immunosuppression: Oral corticosteroids (e.g., prednisone) are initiated three days prior to administration to minimize immune responses, with a gradual taper over the following ten days [38].
- Efficacy Assessment: Functional vision is quantitatively assessed using the Multi-Luminance Mobility Test (MLMT), where patients navigate an obstacle course under varying light conditions (from 400 lux to 1 lux) [38].
Protocol: Systemic rAAV Delivery (Exemplified by Zolgensma)
Zolgensma utilizes a different administration route to target motor neurons.
Key Methodological Details:
- Vector Serotype: AAV9 is selected for its ability to cross the blood-brain barrier after intravenous administration, enabling direct targeting of motor neurons [37].
- Dosing: A one-time infusion is designed to provide lifelong SMN protein expression.
- Outcome Measures: Clinical trials assessed outcomes based on motor milestone achievement (e.g., head control, sitting unassisted) and event-free survival (freedom from permanent ventilation or death) [37].
The Scientist's Toolkit: Essential Reagents for Viral Vector Research
| Research Reagent / Material | Function in Experimental Context | Example Use Case |
|---|---|---|
| AAV Serotype Libraries (e.g., AAV2, AAV5, AAV8, AAV9, AAVrh74) | Determine tissue tropism and transduction efficiency for different target organs. | AAV9 for CNS targets; AAV2 for retinal delivery [37] [39] [38]. |
| Cell-Penetrating Peptides (e.g., Transportan) | Enhance viral vector uptake in difficult-to-transfect cells via bystander uptake and macropinocytosis [44]. | Co-incubation with AAV to improve transfection efficiency in primary retinal pigment epithelium (RPE) cells and macrophages [44]. |
| Minicircle DNA (mcDNA) | A compact, supercoiled DNA vector lacking bacterial sequences, enabling higher and more persistent transgene expression [42]. | Used in non-viral LNP systems (e.g., NCtx) for stable CAR integration in T cells [42]. |
| Ionizable Cationic Lipids (e.g., LP01) | Key component of LNPs; positively charged at low pH to complex with nucleic acids and promote endosomal escape [41]. | Formulated in NTLA-2001 LNP for in vivo CRISPR/Cas9 delivery to the liver [41]. |
| Transposase Systems (e.g., SB100x mRNA) | Enable genomic integration of delivered transgenes for stable long-term expression in dividing cells [42]. | Codelivery with CAR-encoding mcDNA in LNPs to generate persistent in vivo CAR-T cells [42]. |
Viral vectors have demonstrated undeniable success, transitioning from experimental tools to approved medicines for devastating rare diseases. Their proven ability to provide durable therapeutic effects in humans, as evidenced by Zolgensma and Luxturna, sets a high benchmark for any gene delivery system.
The future of gene therapy is not necessarily a choice between viral and non-viral systems, but rather a strategic selection based on the therapeutic objective. rAAVs currently excel in long-term gene replacement strategies, while LNPs offer a versatile platform for transient expression and in vivo genome editing. Emerging innovations, such as the use of ultra-compact CRISPR systems to fit rAAV packaging constraints [39] and targeted LNPs for specific cell types [42], will continue to push the boundaries of both platforms, ultimately expanding the therapeutic arsenal available to researchers and clinicians.
The advent of gene therapy has introduced powerful platforms for treating genetic disorders, cancers, and infectious diseases. The clinical success of these therapies hinges on the delivery vectors that transport genetic material into target cells. Lipid nanoparticles (LNPs) and viral vectors represent the two most prominent delivery technologies, each with distinct advantages and limitations. While viral vectors like lentiviruses and adeno-associated viruses (AAVs) offer high transduction efficiency and potential long-term gene expression, they face challenges related to immunogenicity, pre-existing immunity, and insertional mutagenesis risks [3]. LNPs, clinically validated by mRNA COVID-19 vaccines, provide a non-viral alternative with favorable safety profiles, transient expression, and redosing capability [3]. This guide objectively compares the performance of these platforms, focusing on recent clinical breakthroughs that underscore LNPs' transformative role in modern medicine, from prophylactic vaccines to precision gene editing.
Comparative Analysis: LNP vs. Viral Vector Performance
Table 1: Head-to-Head Comparison of Lipid Nanoparticles (LNPs) and Viral Vectors
| Performance Characteristic | Lipid Nanoparticles (LNPs) | Viral Vectors (Lentivirus, AAV) |
|---|---|---|
| Mechanism of Action | Fuse with cell membrane to deliver payload directly to cytoplasm [3] | Infect cells; may integrate into host genome (lentivirus) or remain episomal (AAV) [3] |
| Typical Gene Expression | Transient (ideal for vaccines, short-term therapies) [3] | Long-term or permanent (ideal for correcting genetic defects) [3] |
| Immune Response | Lower immunogenicity; suitable for repeated dosing [3] [45] | Often triggers stronger immune response; limits redosing potential [3] |
| Delivery Efficiency | High for systemic delivery, but tissue targeting requires engineering [3] [46] | Naturally high efficiency; can be engineered for precise tissue targeting [3] |
| Manufacturing & Scalability | Relatively easy to scale; commercially viable production [3] | Complex and costly large-scale production [3] |
| Key Safety Concerns | Generally strong safety profile; lipid composition must be optimized to minimize toxicity [3] | Risk of insertional mutagenesis and immune reactions [3] |
| Therapeutic Examples | COVID-19 mRNA vaccines, NTLA-2001 (CRISPR for ATTR amyloidosis) [3] [45] | CAR-T cell therapies (Kymriah, Yescarta), Luxturna (retinal disease) [47] [48] |
Table 2: Quantitative Clinical and Preclinical Data for LNP-Based Therapies
| Therapeutic Application | LNP Formulation / Developer | Key Experimental Model & Dose | Reported Efficacy Outcome | Reported Safety Outcome |
|---|---|---|---|---|
| CRISPR for ATTR Amyloidosis (NTLA-2001) | Intellia Therapeutics LNP [45] | Human Phase 1 Trial: Single 55 mg dose | ~90% median reduction in serum TTR at Day 28 [45] | Generally well tolerated; mild infusion-related reaction in 1 of 3 patients upon redosing [45] |
| In vivo CAR-T for B-cell Leukemia | NCtx LNP (anti-CD7/CD3 targeted) [49] | Murine Xenograft Model: Single IV dose | Robust CAR-T cell generation, effective tumor control, and significantly improved survival [49] | Efficient & specific; no major adverse events reported in the study [49] |
| Prophylactic Vaccine (Novel Lipids) | Acuitas Next-Gen LNPs [46] | Preclinical Model: Intramuscular injection | Novel lipids induced equivalent neutralizing antibody titers at a 5-fold lower dose than benchmark ALC-315 [46] | Favorable reactogenicity profiles comparable to ALC-315 [46] |
| Cancer Vaccine | ALC-315 LNP (unmodified mRNA) [46] | Preclinical Model: Intramuscular vs. IV lipoplexes | Induced stronger antigen-specific CD8 T-cell response vs. modified mRNA; equal/superior immunity at one-tenth the dose vs. lipoplexes [46] | Not specified in source, but platform is clinically validated [46] |
Detailed Experimental Protocols for Key LNP Breakthroughs
Protocol: In Vivo Generation of CAR-T Cells Using Targeted LNPs
This protocol is adapted from a study demonstrating the use of targeted LNPs for in vivo CAR-T cell generation, a significant advancement over traditional ex vivo manufacturing [49].
- 1. LNP Formulation and Functionalization: The novel lipid nanoparticle, termed NCtx, is formulated to encapsulate two genetic payloads: a minicircle DNA (mcDNA) encoding the CAR construct and mRNA encoding the SB100x transposase. The LNP surface is then functionalized with T cell-specific targeting ligands, such as anti-CD7 and anti-CD3 binders, to achieve specificity [49].
- 2. In Vitro Specificity and Transfection Efficiency: Primary T cells are isolated and incubated with the NCtx formulation. Specificity is evaluated using flow cytometry to confirm preferential binding and uptake by T cells. Transfection efficiency is assessed by measuring CAR expression via flow cytometry and functionality through antigen-specific cytotoxicity assays and cytokine release profiles [49].
- 3. In Vivo Efficacy in Xenograft Models: Immunodeficient mice are humanized by engrafting them with peripheral blood mononuclear cells (PBMCs) or CD34+ stem cells. These mice are then inoculated with B-cell leukemia cells to establish tumors. A single intravenous dose of NCtx is administered. Tumor growth is monitored over time, and mouse survival is tracked. Blood and tissue samples are analyzed to quantify the generation of CAR-T cells and their persistence in vivo [49].
Protocol: Clinical Proof-of-Concept for CRISPR Redosing
This protocol details the methodology from the first-ever clinical demonstration of redosing an in vivo CRISPR-based therapy, highlighting a key advantage of LNP delivery [45].
- 1. Patient Enrollment and Initial Dosing: In the Phase 1 dose-escalation study for NTLA-2001, three patients with hereditary transthyretin (ATTR) amyloidosis received an initial low dose of 0.1 mg/kg of the investigational therapy. This dose led to a median 52% reduction in serum TTR protein at day 28, which was lower than the target effect [45].
- 2. Follow-on Dosing and Monitoring: After completing the protocol-specified two-year observation period, the same three patients were offered and received a follow-on intravenous dose of NTLA-2001 at 55 mg. Serum TTR levels were measured again at day 28 post-administration. The patients were closely monitored for adverse events, with a specific focus on infusion-related reactions and other potential side effects [45].
- 3. Data Analysis: The pharmacodynamic effect was calculated as the percentage reduction in serum TTR from the original baseline (pre-first dose) and from the pre-second dose baseline. Safety and tolerability data from the redosing event were compared to the safety profile observed after single doses in other trial participants [45].
Visualization of Workflows and Signaling Pathways
LNP-Mediated In Vivo CAR-T Cell Engineering Workflow
This diagram illustrates the multi-step mechanism by which a single intravenous dose of targeted LNPs can generate functional CAR-T cells directly within the patient's body, bypassing complex ex vivo manufacturing.
LNP Redosing Advantage in CRISPR Therapy
This flowchart contrasts the redosing limitations of viral vectors with the flexible dosing potential of LNP-based in vivo therapies, a key differentiator in clinical application.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for LNP-Based Cell Therapy Research
| Reagent / Material | Function in Experimental Protocol |
|---|---|
| Ionizable Lipids (e.g., ALC-315 [46], proprietary blends) | Core component of LNP formulation; enables efficient encapsulation of nucleic acids and endosomal escape for payload release into the cytoplasm. |
| Minicircle DNA (mcDNA) [49] | Engineered, circular DNA vector lacking bacterial sequences; used as a payload to enable prolonged transgene (e.g., CAR) expression. |
| Transposase mRNA (e.g., SB100x [49]) | Co-delivered mRNA payload that encodes an enzyme to facilitate genomic integration of the CAR transgene, leading to stable CAR expression in T cells. |
| T Cell-Targeting Ligands (e.g., anti-CD3, anti-CD7 [49]) | Antibodies or binders conjugated to the LNP surface to confer specificity for T cells, enabling selective targeting and uptake in a complex biological environment. |
| Unmodified mRNA [46] | mRNA payload without nucleoside modification; can induce a stronger CD8+ T-cell response in cancer vaccine applications compared to modified mRNA. |
| Pre-formed Vesicles (PFV) [46] | An alternative LNP manufacturing component that offers equivalent potency with potential improvements in cost, storage, and flexibility for personalized therapies. |
The direct comparison of performance data and experimental evidence solidifies the position of LNPs as a versatile and powerful platform alongside viral vectors. The breakthroughs in CRISPR therapy redosing and in vivo CAR-T cell generation, enabled by LNPs, are not incremental improvements but paradigm shifts. They demonstrate a move away from complex, individualized ex vivo manufacturing towards simpler, more scalable in vivo treatments. While viral vectors remain the preferred choice for therapies requiring lifelong gene expression, the unique advantages of LNPs—including their favorable safety profile, redosing capability, and potential for targeted delivery—are opening new therapeutic frontiers. The ongoing innovation in LNP technology, exemplified by next-generation lipids with enhanced potency and tissue-specific targeting, promises to further expand the reach of genetic medicine.
Gene delivery technologies are pivotal for advancing therapies in oncology, metabolic disorders, and respiratory diseases. Lipid nanoparticles (LNPs) and viral vectors represent two leading platforms, each with distinct advantages and limitations. This guide objectively compares their performance across key applications, supported by experimental data and methodologies.
Lipid Nanoparticles (LNPs) are synthetic carriers composed of ionizable lipids, phospholipids, cholesterol, and PEG-lipids. They encapsulate nucleic acids (e.g., mRNA, siRNA) and facilitate cellular uptake via endocytosis. Their modular design enables rapid production and customization, as demonstrated in mRNA vaccines for COVID-19 [50] [51].
Viral Vectors leverage engineered viruses (e.g., adenoviruses, adeno-associated viruses (AAVs), lentiviruses) to deliver genetic material. They offer high transduction efficiency and durable transgene expression but face challenges such as immunogenicity and limited cargo capacity [52] [53].
Key Differentiators:
- Safety: LNPs avoid pre-existing immunity concerns, while viral vectors may trigger immune responses [52] [53].
- Cargo Flexibility: LNPs accommodate diverse nucleic acids; viral vectors are constrained by genome size (e.g., AAV: <4 kb) [54] [53].
- Manufacturing: LNPs are synthesized via scalable microfluidics; viral vectors require complex biological production [50] [52].
Comparative Performance in Disease Applications
Table 1: Oncology Applications
| Metric | LNP Platform | Viral Vector Platform |
|---|---|---|
| Target | Tumor antigens, immune modulators | Tumor suppressors, oncolytic genes |
| Delivery Efficiency | ~50% tumor regression in melanoma models [51] | ~70% tumor regression in glioma models [53] |
| Key Study | siRNA targeting oncogenes via LNP [51] | Adenovirus-encoded IL-12 [53] |
| Experimental Readout | IFN-γ elevation; T-cell activation [51] | Cytotoxic T-cell infiltration [53] |
Experimental Protocol (Oncology):
- LNP Workflow:
- Formulate LNPs with siRNA targeting PD-L1 using microfluidics.
- Inject intravenously into murine melanoma models (dose: 0.5 mg/kg).
- Assess tumor volume biweekly and quantify T-cells via flow cytometry.
- Viral Vector Workflow:
- Engineer adenovirus encoding IL-12.
- Inject intratumorally into glioma-bearing mice.
- Monitor survival and immune cell infiltration via immunohistochemistry.
Table 2: Metabolic Disorders
| Metric | LNP Platform | Viral Vector Platform |
|---|---|---|
| Target | Hepatocytes for enzyme replacement | Hepatocytes for gene correction |
| Delivery Efficiency | ~30% protein correction in murine liver [51] | ~60% protein correction in hemophilia models [55] |
| Key Study | mRNA encoding factor IX [51] | AAV8-delivered factor VIII [55] |
| Experimental Readout | Plasma factor IX levels; clotting time [51] | Bleeding time reduction [55] |
Experimental Protocol (Metabolic Disorders):
- LNP Workflow:
- Prepare LNPs with mRNA encoding human factor IX.
- Administer intravenously to factor IX-deficient mice.
- Measure factor IX expression via ELISA and clotting assays.
- Viral Vector Workflow:
- Package AAV8 with a codon-optimized factor VIII transgene.
- Inject intravenously into hemophilic mice.
- Quantify factor VIII activity and monitor bleeding phenotypes.
Table 3: Respiratory Diseases
| Metric | LNP Platform | Viral Vector Platform |
|---|---|---|
| Target | Airway epithelium for CFTR mRNA delivery | Lung epithelium for CFTR DNA delivery |
| Delivery Efficiency | ~15% CFTR function restoration in human ALI cultures [56] | ~25% CFTR correction in CF models [54] |
| Key Study | mRNA-LNPs for cystic fibrosis [56] | Lentiviral vectors for CFTR [54] |
| Experimental Readout | Transepithelial chloride current [56] | Mucociliary clearance improvement [54] |
Experimental Protocol (Respiratory Diseases):
- LNP Workflow:
- Formulate LNPs with CFTR mRNA using ionizable lipids (e.g., DLin-MC3-DMA).
- Apply to human bronchial epithelial cultures (ALI model).
- Measure chloride efflux using Ussing chambers.
- Viral Vector Workflow:
- Pseudotype lentivirus with Ebola glycoprotein for apical airway transduction.
- Infect CF patient-derived airway organoids.
- Assess CFTR function via fluorescence-based assays.
Mechanisms of Action and Workflows
Diagram 1: LNP Gene Delivery Mechanism
Title: LNP-Mediated Gene Delivery Pathway
Diagram 2: Viral Vector Gene Delivery Mechanism
Title: Viral Vector-Mediated Gene Delivery Pathway
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for Gene Delivery Research
| Reagent | Function | Example Use Case |
|---|---|---|
| Ionizable Lipids | Encapsulate nucleic acids; enable endosomal escape | DLin-MC3-DMA for mRNA delivery [50] |
| PEG-Lipids | Stabilize LNPs; reduce immunogenicity | DMG-PEG2000 in Onpattro [50] |
| AAV Serotypes | Determine tissue tropism (e.g., AAV8 for liver) | AAV9 for neuronal targeting [53] |
| CRISPR-Cas9 Components | Enable gene editing | LNP-delivered Cas9 mRNA for in vivo editing [51] |
| Transduction Enhancers | Improve viral vector uptake | Poloxmers for adenovirus transduction [54] |
Discussion and Future Perspectives
LNPs excel in safety and rapid development, particularly for mRNA-based therapies, but face challenges in endosomal escape and targeted delivery [51] [57]. Viral vectors offer sustained expression and high efficiency but are limited by immunogenicity and cargo constraints [52] [54]. Emerging solutions include:
- LNP Optimization: Organ-specific targeting via novel lipid designs [57].
- Viral Vector Engineering: Capsid modification to evade immune responses [55].
Future work should focus on combining strengths of both platforms—e.g., LNPs for CRISPR cargo delivery or hybrid systems for personalized oncology vaccines.
The field of gene therapy is navigating a pivotal transition, with lipid nanoparticles (LNPs) emerging as a formidable challenger to the long-standing dominance of viral vectors. While viral vectors such as adeno-associated viruses (AAVs) and lentiviruses have enabled landmark therapies like Luxturna for retinal dystrophy and Zolgensma for spinal muscular atrophy, concerns regarding their immunogenicity, potential for insertional mutagenesis, and challenges with redosing have prompted the exploration of safer, more versatile alternatives [1] [11]. LNPs, validated by their successful deployment in COVID-19 mRNA vaccines, offer a promising non-viral platform characterized by lower immunogenicity, a superior payload capacity for complex cargo like CRISPR-Cas9 components, and greater flexibility for repeated administrations [3] [58]. The administration route is a critical factor in the success of gene therapies, and the intranasal and pulmonary pathways represent particularly promising frontiers for LNP delivery, offering direct access to the lungs for treating local diseases and potential entry points to the central nervous system [59] [60].
Core Technology Comparison
Fundamental Characteristics and Clinical Applications
The choice between viral vectors and LNPs involves a fundamental trade-off between the durable expression offered by viruses and the safety and flexibility of synthetic nanoparticles. The table below summarizes their core characteristics and clinical status.
Table 1: Fundamental Characteristics of Viral Vectors and Lipid Nanoparticles
| Feature | Viral Vectors (e.g., AAV, Lentivirus) | Lipid Nanoparticles (LNPs) |
|---|---|---|
| Genetic Material Delivered | Primarily DNA [1] | mRNA, siRNA, DNA, CRISPR-Cas9 components [59] [3] |
| Mechanism of Action | Cell entry via viral receptors; some types integrate into host genome [1] | Fusion with cell membrane; payload released into cytoplasm [3] |
| Typical Expression Duration | Long-term to permanent (e.g., with AAVs and Lentiviruses) [3] | Transient (ideal for vaccines, short-term protein expression) [3] |
| Immunogenicity | Often high; pre-existing immunity and strong immune response post-administration can limit redosing [1] [11] | Generally lower, allowing for repeated dosing [3] [58] |
| Payload Capacity | Limited (e.g., AAV: ~4.7 kb) [11] | Larger (can deliver payloads up to 10 kb) [58] |
| Key Safety Concerns | Insertional mutagenesis, strong immune responses (e.g., liver toxicity) [1] [11] | Potential toxicity related to lipid composition; generally considered safer [3] [58] |
| Scalability & Manufacturing | Complex and costly to manufacture at scale [3] [11] | Relatively easier and more cost-effective to scale [3] [58] |
| Representative Approved Therapies | Luxturna (AAV), Zolgensma (AAV), Strimvelis (Retrovirus) [1] [48] | Onpattro (LNP-siRNA), Comirnaty (LNP-mRNA), Spikevax (LNP-mRNA) [59] [60] |
Performance in Intranasal and Pulmonary Delivery
When applied to respiratory delivery, both platforms face unique challenges. A significant hurdle for inhaled LNPs is maintaining stability during nebulization, a process that can cause disintegration and payload leakage [61]. Viral vectors, while efficient at transfection, face formidable immune system barriers. The table below compares their performance for inhalation.
Table 2: Performance in Intranasal and Pulmonary Delivery Applications
| Aspect | Viral Vectors | Lipid Nanoparticles (LNPs) |
|---|---|---|
| Primary Target Cells in Lungs | Respiratory epithelial cells (with Adenoviruses) [1] | Dendritic cells, epithelial cells; target cell can be modulated by formulation [59] [61] |
| Key Challenge in Inhalation | Pre-existing and treatment-induced immune responses; potential for inflammation [11] | Physical instability during nebulization (shear force-induced disintegration) [59] [61] |
| Redosing Capability for Chronic Lung Diseases | Severely limited due to immune response [11] | Feasible due to lower immunogenicity [3] [58] |
| Clinical Stage for Respiratory Disease | Clinical trials for cystic fibrosis (e.g., AAV-CFTR) [1] | Clinical trials for cystic fibrosis (e.g., ARCT-032, MRT5005) [59] |
| Notable Preclinical/Clinical Outcomes | Phase I study of AAV-CFTR in CF patients demonstrated safety but limited efficacy [1] | Inhaled CAS-LNP in large animals (dogs, pigs) elicited robust mucosal immunity; LOOP-LNP platform showed significant fibrosis inhibition in mice [61] [62] |
Advanced LNP Formulation Strategies for Inhalation
To overcome the instability of LNPs during nebulization, researchers have developed innovative formulation strategies. Two notable advanced approaches are the Charge-Assisted Stabilization (CAS) strategy and the "LOOP" screening platform.
Charge-Assisted Stabilization (CAS) Strategy
The CAS strategy addresses the instability of traditional LNPs by introducing electrostatic repulsions between particles. Researchers integrated a negatively charged peptide-lipid conjugate (DSSC-DOPE) into the standard LNP formulation. This modification conferred a stable negative surface charge (zeta-potential of ~ -17 mV for the 2.5% CAS-LNP) without affecting size or encapsulation efficiency. The resulting CAS-LNP demonstrated exceptional stability during nebulization, with a much higher percentage of intact particles compared to traditional LNPs, leading to efficient mRNA delivery to dendritic cells in the lungs and robust immune responses in large animal models [61].
The "LOOP" Screening Platform
The LOOP platform is a systematic, four-step workflow designed to screen and optimize inhaled LNPs (iLNPs) for superior stability and protein expression [62]:
- Lipid Ratio Adjustment: Using microfluidics to prepare a library of LNPs with varying molar ratios of ionizable lipids, helper lipids, cholesterol, and PEG-lipids.
- Dialysate Optimization: Dialyzing LNPs against buffers of different compositions and pH (e.g., HEPES pH 6.0 was found optimal).
- Nebulization Buffer Enhancement: Introducing excipients like ethanol, propylene glycol, or poloxamer 188 into the nebulization buffer to enhance colloidal stability against shear forces.
- Iterative Re-optimization: Re-adjusting lipid ratios based on the new dialysis and nebulization buffers to investigate the "lipid composition-activity" relationship. This platform enabled the development of iLNP-HP08LOOP, which, when loaded with mRNA encoding an anti-fibrotic protein (IL-11 scFv), significantly inhibited pulmonary fibrosis in mice and outperformed intravenously injected therapeutics [62].
Experimental Workflows and Visualization
Workflow for Developing Stable iLNPs
The following diagram synthesizes the key steps from both the CAS and LOOP strategies into a coherent workflow for developing stable, effective inhaled LNPs.
Diagram 1: Workflow for stable inhaled LNP development.
Signaling Pathway for LNP-Mediated mRNA Delivery
The mechanism by which inhaled LNPs deliver their mRNA payload into target cells involves a defined intracellular pathway, which is visualized below.
Diagram 2: Mechanism of LNP-mediated mRNA delivery.
The Scientist's Toolkit: Essential Research Reagents
The development and testing of inhaled LNP formulations rely on a specific set of reagents, devices, and biological models. The following table catalogues key solutions used in the featured research.
Table 3: Essential Research Reagents and Materials for Inhaled LNP Studies
| Reagent / Material | Function / Role | Specific Examples / Notes |
|---|---|---|
| Ionizable Lipids | Critical for mRNA encapsulation and endosomal escape; positively charged at low pH [59]. | SM-102 (Moderna's choice), ALC-0315 (Pfizer's choice), DLin-MC3-DMA (Onpattro), proprietary compounds like AA3-DLin [59] [62]. |
| Helper Lipids | Stabilize the LNP bilayer structure and influence membrane fusion properties [59]. | DSPC, DOPE. DOPE can be conjugated to peptides (e.g., DSSC-DOPE) for charge stabilization [61]. |
| PEG-Lipids | Stabilize LNPs during formation, prevent aggregation, influence pharmacokinetics [59]. | DMG-PEG2000. Amount must be optimized as high content can hinder cellular uptake [59] [61]. |
| Stabilizing Excipients | Enhance colloidal stability of LNPs during the shear stress of nebulization [62]. | Ethanol, Propylene Glycol, Poloxamer 188 (used in the LOOP platform nebulization buffer) [62]. |
| Optimized Dialysis Buffers | Post-assembly processing to create a stable final formulation environment [62]. | HEPES buffer at pH 6.0 was identified as optimal in the LOOP platform [62]. |
| Nebulizer Device | Aerosolizes LNP solution into micron-sized droplets for deep lung deposition [59] [60]. | Vibrating mesh nebulizers (e.g., Aerogen Solo) are commonly used in research for efficiency [61]. |
| Reporter mRNA | Encodes a easily detectable protein to screen and quantify LNP delivery efficiency in vitro and in vivo. | mRNA encoding firefly luciferase (mFluc) or other reporter genes (e.g., mLuc) [61] [62]. |
| Animal Disease Models | Preclinical in vivo testing for efficacy and toxicity of the LNP therapeutic. | Mouse models of pulmonary fibrosis (e.g., bleomycin-induced), metastatic lung cancer, or infection (e.g., SARS-CoV-2) [61] [62]. |
Intranasal and pulmonary delivery of LNPs represents a rapidly advancing frontier in gene therapy, offering a potent and versatile platform for tackling respiratory diseases, enabling mucosal vaccination, and potentially accessing the central nervous system. While viral vectors remain a powerful tool for certain applications requiring long-term gene expression, LNPs present a compelling alternative with distinct advantages in safety, manufacturing scalability, and redosing capability—critically important attributes for chronic respiratory conditions. The innovative formulation strategies, such as charge-assisted stabilization and systematic screening platforms, are decisively overcoming previous technical hurdles like nebulization instability. As these technologies mature and converge, inhaled LNP-based gene therapies are poised to transition from promising preclinical results to transformative clinical realities, ultimately offering new hope for patients with a wide spectrum of intractable lung diseases.
Navigating Technical Hurdles and Enhancing Performance
The success of gene therapies and vaccines is critically dependent on the delivery vehicle that transports genetic cargo into target cells. Among the most prominent platforms are viral vectors and lipid nanoparticles (LNPs), each presenting distinct immunological challenges that can impact both safety and efficacy. Immunogenicity—the ability of a substance to provoke an immune response—represents a significant hurdle for both viral and non-viral delivery systems. For viral vectors, preexisting immunity in human populations can neutralize the vector before it delivers its therapeutic payload. For LNPs, the components of the nanoparticle itself can trigger innate immune responses that may lead to adverse effects or reduce therapeutic effectiveness. Understanding these immune reactions is paramount for developing next-generation gene therapies that are both safe and effective [3] [63].
The immunological profiles of these two platforms differ substantially. Viral vectors, particularly those based on common human pathogens like adenoviruses, often encounter preexisting neutralizing antibodies in patient populations. Conversely, LNP immunogenicity primarily stems from activation of the innate immune system through pattern recognition receptors, which can lead to inflammatory responses and, in some cases, impact adaptive immunity. This review systematically compares the immunogenicity challenges associated with viral vectors and lipid nanoparticles, examining the mechanisms of immune activation, strategies to overcome these limitations, and experimental approaches for characterizing immune responses to guide the development of safer gene delivery platforms [3] [63] [64].
Comparative Immunogenicity Profiles: Viral Vectors vs. Lipid Nanoparticles
Mechanisms of Immune Activation
Viral vectors and lipid nanoparticles activate the immune system through fundamentally different pathways, each with distinct implications for therapeutic applications.
Table 1: Comparative Immunogenicity Profiles of Viral Vectors and Lipid Nanoparticles
| Feature | Viral Vectors | Lipid Nanoparticles |
|---|---|---|
| Primary Immune Trigger | Viral capsid proteins and viral genetic material [3] | Ionizable lipids, PEG-lipids, and mRNA component [63] [64] |
| Preexisting Immunity | Common; significant portion of population has neutralizing antibodies [3] | Limited; no preexisting antibodies to LNPs themselves [3] |
| Primary Immune Response | Adaptive immune response; antibody-mediated neutralization [3] | Innate immune response; inflammation and cytokine production [63] |
| Repeat Dosing Potential | Limited due to enhanced neutralization upon redosing [3] | More feasible, though anti-PEG antibodies may develop [3] [63] |
| Key Immune Sensors | Antibodies, T-cell receptors [3] | TLRs, RLRs, NLRs (particularly TLR7/8 for mRNA) [63] [64] |
| Typical Onset | Days to weeks (adaptive immunity) [3] | Hours (innate immunity) [63] [64] |
Viral vectors, particularly those derived from adenoviruses, are highly efficient at gene delivery but face a substantial challenge from preexisting immunity. Because many viral vectors are based on common human pathogens, a significant proportion of the population possesses neutralizing antibodies that can bind to the viral capsid and prevent cellular entry. This effectively reduces the therapeutic dose that reaches target cells and can limit treatment efficacy. Additionally, administration of viral vectors typically triggers a robust adaptive immune response that generates long-lasting memory, making repeated administrations increasingly ineffective as the immune system rapidly clears subsequent doses [3].
In contrast, lipid nanoparticles primarily stimulate the innate immune system through various pattern recognition receptors (PRRs). The LNP structure itself can act as a foreign material that immune cells recognize as potentially dangerous. Furthermore, the genetic payload within LNPs—particularly mRNA—can be detected by intracellular sensors like Toll-like receptors (TLR7/8) and RIG-I-like receptors (RLRs), triggering signaling cascades that produce type I interferons and proinflammatory cytokines. Recent research has demonstrated that the mRNA component itself, rather than just the LNP structure, is essential for inducing a potent innate immune response characterized by rapid activation of dendritic cells and recruitment of monocytes to draining lymph nodes [63] [64].
Impact on Therapeutic Efficacy and Safety
The immunogenicity of both delivery systems has direct implications for their therapeutic utility. For viral vectors, preexisting immunity can dramatically reduce treatment efficacy. For instance, in gene therapy trials using adenovirus vectors, patients with preexisting antibodies showed significantly reduced transgene expression compared to seronegative individuals. This limitation has prompted the development of alternative viral vectors based on less common serotypes or non-human viruses, though these too may eventually encounter immunity issues with repeated use [3].
For LNPs, immune activation presents a more complex picture. While excessive inflammation can lead to adverse effects like fever, fatigue, and injection site pain, moderate immune stimulation can actually be beneficial for vaccine applications by providing adjuvant activity. However, for non-vaccine applications such as protein replacement therapies or gene editing, immune activation is generally undesirable as it can reduce protein expression and potentially exacerbate underlying conditions. Studies have shown that type I interferon responses induced by LNP-mRNA vaccines can paradoxically attenuate adaptive immunity by suppressing the translation of the encoded antigen, highlighting the complex interplay between innate immune activation and therapeutic efficacy [64].
Experimental Approaches for Characterizing Immune Responses
Assessing LNP Immunogenicity
The evaluation of LNP-induced immunogenicity requires a multifaceted approach that examines both the components of the nanoparticle and their interactions with immune cells. Standard experimental protocols include:
In vitro immune cell activation assays: Isolated human peripheral blood mononuclear cells (PBMCs) or specific immune cell subsets (e.g., dendritic cells, monocytes) are exposed to LNPs or their individual components. Activation is measured through surface marker expression (e.g., CD80, CD86, MHC-II via flow cytometry) and cytokine production (e.g., IL-6, TNF-α, IFNs via ELISA or multiplex assays) after 24-48 hours of exposure. Dose-response curves help establish the potency of immune activation [63] [64].
PRR signaling pathway assays: To identify specific pattern recognition receptors involved in LNP recognition, reporter cell lines expressing individual PRRs (e.g., TLR7, TLR8, RIG-I) are employed. These cells contain inducible luciferase or SEAP (secreted embryonic alkaline phosphatase) genes under the control of promoters responsive to PRR activation (NF-κB or IRF pathways). Cells are exposed to LNPs, and receptor activation is quantified by measuring luminescence or SEAP activity, allowing researchers to pinpoint which sensors are engaged by the delivery system [65] [63].
Animal models for in vivo immunogenicity: Mice are commonly used to evaluate LNP immunogenicity, with C57BL/6 and BALB/c strains being most frequent. Animals receive LNP formulations via relevant routes (e.g., intramuscular, intravenous), and immune parameters are assessed at multiple time points. Key measurements include serum cytokine levels, immune cell recruitment to injection sites and draining lymph nodes, and antigen-specific antibody responses. Transgenic models, such as IFNAR-/- mice lacking type I interferon receptors, help elucidate specific signaling pathways involved in the immune response [64].
Table 2: Key Immune Parameters for Assessing LNP Immunogenicity
| Parameter Category | Specific Measurements | Methodology | Significance |
|---|---|---|---|
| Innate Immunity | Type I interferon levels (IFN-α/β), Proinflammatory cytokines (IL-6, TNF-α) | ELISA, multiplex immunoassays | Measures acute inflammatory response [63] [64] |
| Immune Cell Activation | Surface marker expression (CD80, CD86, MHC-II), Dendritic cell maturation | Flow cytometry, immunostaining | Indicates antigen-presenting cell activation [63] |
| PRR Engagement | NF-κB activation, IRF pathway activation | Reporter assays, Western blot | Identifies specific sensing pathways [65] [63] |
| Adaptive Immunity | Antigen-specific antibodies (IgG, subtypes), T-cell responses (IFN-γ ELISpot) | ELISA, ELISpot, intracellular staining | Measures functional immune activation [64] |
Evaluating Preexisting Immunity to Viral Vectors
Assessing preexisting immunity to viral vectors is crucial for predicting therapeutic efficacy and selecting appropriate patient populations. Standard approaches include:
Neutralization assays: Serum from patients is incubated with the viral vector, and the mixture is applied to permissive cells in culture. The reduction in transduction efficiency, typically measured by reporter gene expression (e.g., GFP, luciferase), indicates the level of neutralizing antibodies present. The neutralization titer is defined as the serum dilution that reduces transduction by 50% (NT50) or 90% (NT90) compared to control sera. These assays help establish cutoff values for patient inclusion in clinical trials [3].
ELISA for anti-vector antibodies: High-throughput enzyme-linked immunosorbent assays (ELISAs) are used to detect and quantify anti-capsid antibodies in patient sera. Purified viral capsid proteins are coated onto plates, patient serum is applied, and bound antibodies are detected using enzyme-conjugated secondary antibodies. This approach provides a quantitative measure of total antibody binding but does not distinguish between neutralizing and non-neutralizing antibodies [3].
T-cell assays: The presence of capsid-specific T cells can also impact viral vector efficacy and safety. IFN-γ ELISpot or intracellular cytokine staining assays measure T-cell responses to viral vector antigens. These responses may contribute to vector clearance and potentially lead to immunotoxicities, making their assessment clinically relevant [3].
Strategies to Overcome Immunogenicity Challenges
Engineering Less Immunogenic Viral Vectors
Several innovative approaches have been developed to circumvent preexisting immunity to viral vectors:
Vector serotype switching and engineering: Researchers have developed viral vectors based on rare human serotypes or non-human viruses that have lower seroprevalence in human populations. For example, adenovirus vectors derived from chimpanzee viruses have shown promise in clinical trials. Additionally, structural engineering of viral capsids can modify or shield epitopes targeted by neutralizing antibodies while maintaining receptor binding capability. This "vector shuffling" approach creates chimeric vectors with reduced cross-reactivity to common human serotypes [3].
Immunosuppression and vector masking: Transient immunosuppression with drugs such as corticosteroids, cyclosporine, or monoclonal antibodies can temporarily blunt immune responses against viral vectors, allowing for more efficient transduction. Alternatively, physically shielding viral vectors with polymers like polyethylene glycol (PEG) can reduce antibody recognition, though this approach must balance stealth properties with maintained infectivity [3].
Modulating LNP-Induced Immune Responses
The immunogenicity of LNPs can be modulated through both formulation optimization and component engineering:
mRNA engineering: Nucleoside modification, particularly the replacement of uridine with N1-methylpseudouridine, significantly reduces recognition by innate immune sensors like TLR7/8 while enhancing protein expression. Additionally, careful purification of mRNA to remove double-stranded RNA (dsRNA) contaminants minimizes activation of sensors such as MDA5 and PKR. These modifications collectively create "immunologically silent" mRNA that evades detection while maintaining high translational efficiency [64].
Lipid component optimization: The structure of ionizable lipids significantly influences LNP immunogenicity. Saturated hydrocarbon chains tend to be less inflammatory than their unsaturated counterparts. Similarly, reducing PEG-lipid content or using alternative PEG-lipid structures can minimize anti-PEG immune responses while maintaining nanoparticle stability. Some advanced LNP formulations incorporate immune modulators, such as TLR antagonists, directly into the nanoparticle to actively suppress immune recognition [63] [64].
Incorporation of immunomodulators: Recent research has demonstrated that incorporating specific immune agonists can strategically enhance LNP immunogenicity for vaccine applications. A 2024 study showed that replacing a small portion (0.5%) of cholesterol in LNPs with a TLR7/8 agonist significantly enhanced both cellular and humoral immune responses against various antigens, including HPV and SARS-CoV-2. This approach demonstrates how rational design can harness immune activation for therapeutic benefit while maintaining acceptable safety profiles [65].
Signaling Pathways in LNP-Mediated Immune Activation
The innate immune response to lipid nanoparticles involves multiple interconnected signaling pathways that recognize both the LNP structure and its nucleic acid payload. Understanding these pathways is essential for developing strategies to control immunogenicity.
LNP Immune Signaling Pathways
The diagram above illustrates the key signaling pathways involved in immune recognition of lipid nanoparticles. The mRNA component is primarily detected by endosomal TLR7/8 and cytosolic RIG-I/MDA5, leading to NF-κB and IRF activation that drives inflammatory cytokine and type I interferon production. Simultaneously, the LNP structure itself can activate the NLRP3 inflammasome, resulting in pyroptosis and IL-1β/IL-18 release. These pathways collectively contribute to dendritic cell activation and the initiation of adaptive immunity, but can also suppress protein translation and contribute to adverse effects [63] [64].
Table 3: Essential Research Reagents for Immunogenicity Studies
| Reagent/Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| TLR Agonists/Antagonists | Resiquimod (R848), Vesatolimod (GS-9620), Imidazoquinoline derivatives [65] | Pathway-specific immune modulation | Define contribution of specific TLRs to LNP immunogenicity [65] |
| Cytokine Detection | ELISA kits (IFN-α, IFN-β, IL-6, TNF-α), Multiplex bead arrays, ELISpot kits [64] | Quantifying immune activation | Measure innate and adaptive immune responses to LNPs [64] |
| Reporter Systems | HEK-Blue hTLR cells, NF-κB luciferase reporters, SEAP reporters [65] | Pathway-specific activation screening | Identify specific PRRs engaged by LNP components [65] |
| Animal Models | C57BL/6, BALB/c, IFNAR-/- mice, Humanized mouse models [64] | In vivo immunogenicity assessment | Evaluate immune responses in physiologically relevant context [64] |
| Ionizable Lipids | ALC-0315, SM-102, DLin-MC3-DMA, Novel synthetic lipids [66] [64] | LNP formulation optimization | Structural components that influence immunogenicity and delivery efficiency [66] [64] |
| mRNA Modifications | N1-methylpseudouridine, 5-methoxyuridine, Purified mRNA [64] | mRNA engineering | Reduce innate immune recognition while enhancing translation [64] |
The immunogenicity of gene delivery vectors represents both a challenge and an opportunity for therapeutic development. Viral vectors face significant hurdles from preexisting immunity that can neutralize the vector before it reaches target cells, while lipid nanoparticles trigger complex innate immune responses that can both enhance and interfere with therapeutic applications. The optimal choice between these platforms depends heavily on the specific application—viral vectors remain advantageous for long-term gene expression in immunologically naive tissues, while LNPs offer superior safety profiles and redosing capability for vaccines and transient therapies.
Future directions in the field include the development of stealth viral vectors with modified capsids that evade neutralizing antibodies, and precision-engineered LNPs with tunable immunogenicity. The incorporation of specific immune modulators directly into LNP formulations represents a promising approach to harness beneficial immune activation while minimizing detrimental inflammation. Additionally, advanced delivery strategies that target specific tissues and cell types may reduce off-target immune activation. As our understanding of the intricate relationships between delivery systems and the immune system deepens, we move closer to the ideal of customizable gene delivery platforms with precisely controlled immunogenicity profiles for diverse therapeutic applications [3] [63] [64].
The success of gene therapy hinges on the precise delivery of genetic cargo to target cells. Two dominant delivery systems, lipid nanoparticles (LNPs) and viral vectors, present researchers with a critical dilemma: how to balance therapeutic efficacy with potential toxicity and manageable production costs. This dosage challenge is multifaceted, influenced by factors from immunogenicity and manufacturing complexity to intracellular processing. The choice between a transient, easily manufactured LNP dose and a potent, long-lasting but complex viral vector dose fundamentally shapes the therapeutic and commercial viability of a gene therapy. This guide provides an objective, data-driven comparison of these platforms to inform research and development strategies.
System Mechanisms and Key Characteristics
Lipid nanoparticles and viral vectors function through fundamentally different biological mechanisms, which directly influence their dosing regimens, safety profiles, and manufacturing processes.
Figure 1: Comparative Gene Delivery Pathways. LNPs and viral vectors navigate distinct intracellular routes to deliver their genetic payload, facing different biological barriers that impact overall efficiency [3] [28].
Lipid Nanoparticles (LNPs)
LNPs are synthetic, spherical carriers that encapsulate RNA. They deliver their payload by fusing with the cell membrane, entering via endocytosis, and releasing their cargo into the cytoplasm through endosomal escape. This process is facilitated by ionizable lipids that become protonated in the acidic endosomal environment, promoting a transition to a hexagonal lipid phase that disrupts the endosomal membrane [3] [28]. This mechanism typically results in transient gene expression, making LNPs ideal for vaccines and short-term therapies. A key limitation is that components of the LNP and its RNA payload can segregate during endosomal sorting, and only a small fraction of the internalized RNA successfully escapes to the cytosol [28].
Viral Vectors
Viral vectors, such as adeno-associated virus (AAV) and lentivirus, are engineered viruses that leverage natural infection pathways. They often enter cells through receptor-mediated endocytosis. AAV vectors predominantly remain as episomal DNA in the nucleus, leading to long-term expression in non-dividing cells. Lentiviral vectors can integrate their genome into the host cell's DNA, potentially enabling permanent gene correction but carrying a risk of insertional mutagenesis [3] [67]. Their high efficiency is partly due to co-evolution with hosts, but their immunogenicity can limit re-dosing [3].
Quantitative System Comparison
The fundamental differences in the mechanisms of LNPs and viral vectors translate into distinct performance profiles. The table below summarizes key characteristics that directly impact the dosage challenge.
Table 1: Performance and Dosage Profile Comparison of Gene Delivery Systems
| Characteristic | Lipid Nanoparticles (LNPs) | Viral Vectors (AAV/Lentivirus) |
|---|---|---|
| Primary Mechanism | Endosomal fusion & escape [3] | Receptor-mediated cell entry [3] [67] |
| Typical Cargo | mRNA, siRNA, CRISPR components [3] | DNA, CRISPR ribonucleoproteins [67] |
| Expression Duration | Transient (days to weeks) [3] | Long-term (months to years) [3] |
| Immunogenicity | Lower; suitable for repeated dosing [3] | Higher; immune response limits re-administration [3] |
| Manufacturing Scalability | Highly scalable (e.g., microfluidics) [3] [68] | Complex and costly; challenges in scalability [3] [69] |
| Tissue Targeting | Improving; predominantly liver-tropic [31] | Precise; can be engineered for specific tissues [3] [33] |
| Key Dosage Advantage | Lower toxicity risk, re-dosable | High single-dose efficacy, sustained effect |
| Key Dosage Limitation | Lower delivery efficiency to cytosol [28] | Immune response, production limits supply [69] |
Manufacturing and Dosage Implications
The path from benchtop to clinic is heavily influenced by the production scalability of the delivery system, which in turn affects the cost and availability of a therapeutic dose.
LNP Manufacturing
LNP production has been revolutionized by microfluidic technologies, which allow for precise control over critical quality attributes like particle size. Studies show that controlling the flow rate in a microfluidic chip can yield LNPs in a tightly controlled size range (e.g., 30–270 nm) while maintaining identical lipid ratios [68]. This is crucial because smaller LNPs (e.g., ~80 nm) have demonstrated higher cellular uptake and transfection efficiency in vitro and altered biodistribution in vivo [68]. The ability to fine-tune size and composition in a scalable process makes LNP manufacturing highly adaptable and easier to scale compared to viral vectors, supporting more consistent dosing and lower production costs at commercial scales.
Viral Vector Manufacturing
Producing viral vectors like AAV involves complex biological systems, such as transfection of mammalian cell cultures. The process faces challenges with variability, low yields, and difficulties in scaling up, which constrains the supply of clinical-grade material [69]. Key bottlenecks include the separation of full capsids (which contain the therapeutic gene) from empty capsids during downstream purification. While innovations in serotype-agnostic affinity chromatography are improving this, the process remains inherently more variable and costly than LNP synthesis [69]. This manufacturing complexity is a primary driver of the high cost of viral vector-based therapies and can limit dose availability.
Experimental Approaches and Protocols
Research into overcoming the dosage challenge relies on robust experimental methods. The following protocols are representative of current approaches to enhance the efficiency of both systems.
Protocol 1: Enhancing Viral Vector Delivery with Transportan Peptide
This method details a simple co-incubation strategy to improve viral transduction, particularly in difficult-to-transfect cells [44].
- Objective: To significantly enhance the transduction efficiency of lentivirus and adeno-associated virus (AAV) vectors.
- Materials:
- Transportan (TP) Peptide: A 27-amino-acid amphiphilic cell-penetrating peptide (Sequence: GWTLNSAGYLLGKINLKALAALAKKIL).
- Viral Vectors: GFP-expressing lentivirus or AAV.
- Cell Culture: Target cells (e.g., difficult-to-transfect cell lines, primary macrophages, or retinal pigment epithelium cells).
- Standard cell culture reagents and equipment.
- Method:
- Preparation: Dilute the GFP-expressing viral vectors and the TP peptide in the appropriate serum-free medium.
- Co-incubation: Simply mix the diluted viral vectors with the TP peptide. The study used varying dilutions of virus and TP concentrations (e.g., 2.5 - 10 μM) [44].
- Transduction: Add the virus-TP mixture directly to the cells and incubate for 48 hours.
- Analysis: Assess transduction efficiency via fluorescent imaging or flow cytometry to quantify GFP-positive cells.
- Mechanism: TP induces a bystander uptake effect, enhancing viral entry into cells through macropinocytosis without requiring a direct chemical bond between the peptide and the virus [44].
Protocol 2: Microfluidic Size-Tuning of LNPs for Optimized Delivery
This protocol describes a simulation-based method to produce LNPs of specific sizes to study and optimize their delivery performance [68].
- Objective: To generate LNPs with controlled size and evaluate its effect on mRNA delivery efficiency in vitro and in vivo.
- Materials:
- Lipids: Ionizable cationic lipid, phospholipid, cholesterol, PEGylated lipid.
- Microfluidic Chip: Designed with computational fluid dynamics (CFD) simulations to achieve a specific mixing index (e.g., 70%).
- mRNA Cargo: e.g., Firefly luciferase or GFP mRNA.
- Syringe pumps, dynamic light scattering (DLS) for size/zeta potential measurement.
- Method:
- Chip Design: Use CFD simulations to model the mixing index within the microfluidic channel and identify total flow rates (TFR) and flow rate ratios (FRR) that achieve the target mixing.
- LNP Formulation: Prepare lipid solutions in ethanol and mRNA in aqueous buffer. Use syringe pumps to introduce both streams into the microfluidic chip at the predetermined TFR and FRR.
- Size Control: Tune the flow rates to produce LNPs across a desired size range (e.g., 30 nm to 270 nm) while keeping the lipid composition identical [68].
- Characterization: Measure the size, polydispersity index (PDI), and zeta potential of the formulated LNPs using DLS.
- Evaluation: Test LNP performance in cellular uptake and gene expression assays in vitro (e.g., HeLa cells) and analyze biodistribution and mRNA expression following intravenous or intramuscular administration in vivo.
- Key Finding: Smaller LNPs (e.g., ~80 nm) within the tested range exhibited higher cellular uptake and transfection efficiency [68].
Advanced Challenges: The Intracellular Barrier
Even after successful cellular uptake, significant barriers remain that limit the efficiency of a delivered dose. Recent research using live-cell microscopy has identified that only a small fraction of internalized LNP cargo actually escapes the endosome to reach the cytosol [28]. Furthermore, segregation can occur between the ionizable lipid and the RNA payload within the endosomal system, and many endosomes damaged by LNPs contain no detectable RNA. These inefficiencies represent major post-uptake bottlenecks, meaning a large portion of the administered dose may never reach its target, necessitating higher initial doses to achieve a therapeutic effect [28].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Gene Delivery System Research and Development
| Reagent / Material | Primary Function | Application Context |
|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs; enables encapsulation and endosomal escape via protonation [70]. | LNP Formulation |
| PEGylated Lipids | Stabilizes LNP formation, controls particle size, modulates pharmacokinetics and cellular uptake [70]. | LNP Formulation |
| Cell-Penetrating Peptides (e.g., Transportan) | Enhates cellular uptake of co-incubated payloads via induction of macropinocytosis [44]. | Viral Vector & Non-Viral Enhancement |
| Microfluidic Devices | Enables reproducible, scalable production of monodisperse LNPs with precise size control [68]. | LNP Manufacturing |
| Adeno-Associated Virus (AAV) Serotypes | Engineered viral capsids with varying tropism for targeting specific tissues (e.g., brain, liver, muscle) [67] [33]. | Viral Vector Delivery |
| Affinity Chromatography Resins | Critical for downstream purification of viral vectors; separates full capsids from empty capsids [69]. | Viral Vector Manufacturing |
The dosage challenge in gene delivery presents a clear trade-off. Lipid nanoparticles offer a scalable, re-dosable platform with a favorable safety profile, well-suited for transient expression needs. However, their current limitations in delivery efficiency and tissue targeting beyond the liver often require higher doses to be effective. Viral vectors provide unparalleled delivery efficiency and long-lasting expression from a single dose, ideal for curing genetic disorders, but are hampered by immunogenicity, complex manufacturing, and higher production costs. The future lies not in choosing a single winner, but in leveraging the strengths of each platform based on the therapeutic application, and in innovating to overcome their respective limitations, ultimately making safe and effective gene therapies more accessible.
The success of modern gene therapy hinges on the precise delivery of genetic cargo to target cells, achieving sustained therapeutic expression while minimizing off-target effects. Two leading technologies—lipid nanoparticles (LNPs) and viral vectors—dominate this landscape, each with distinct advantages and limitations. LNPs, versatile synthetic carriers, excel in delivering a wide range of nucleic acids with lower immunogenicity, facilitating repeat administration. In contrast, viral vectors, engineered from naturally evolved viruses, offer unparalleled delivery efficiency and the potential for long-lasting gene expression, making them suitable for curative intent therapies. This guide provides an objective, data-driven comparison of these platforms, focusing on the critical parameters of expression durability and tissue-specific targeting to inform rational technology selection for research and drug development.
The fundamental difference between LNPs and viral vectors lies in their core design and interaction with target cells. The table below summarizes their primary mechanisms.
Table 1: Core Mechanism Comparison: LNPs vs. Viral Vectors
| Feature | Lipid Nanoparticles (LNPs) | Viral Vectors |
|---|---|---|
| Composition | Synthetic lipids (ionizable, structural, PEG-lipid, cholesterol) [3] [71] | Engineered viral capsid (e.g., AAV, Adenovirus, Lentivirus) [3] [72] |
| Genetic Cargo | mRNA, siRNA, CRISPR-Cas9 components (RNA/DNA) [3] [73] | DNA, with some supporting self-amplifying RNA [72] |
| Delivery Process | Endocytosis → Endosomal escape → Cytosolic release [3] [28] | Cell receptor binding → Endocytosis → Viral uncoating → Nuclear entry [3] [72] |
| Fate of Genetic Material | Remains episomal in cytoplasm; transient expression [3] | Can remain episomal (AAV) or integrate into host genome (Lentivirus); potential for long-term expression [3] [72] |
The following diagram illustrates the distinct intracellular pathways of these two delivery systems.
Quantitative Performance Data
Durability of Gene Expression
The duration of therapeutic gene expression is a critical differentiator, dictated by the underlying delivery mechanism.
Table 2: Durability and Expression Profile Comparison
| Platform | Typical Expression Duration | Key Determinants | Supporting Data |
|---|---|---|---|
| LNP | Transient (Days to Weeks) [3] | mRNA half-life; non-integrating; LNP stability. | Ideal for vaccines, short-term protein production [3]. |
| Viral Vector | Long-term to Permanent (Months to Years) [3] | Vector serotype (AAV: episomal; Lentivirus: integrating); target cell division rate. | Hemophilia B (Etranacogene dezaparvovec): Mean FIX activity of 37.4% sustained at 4 years post-treatment [74]. AAV Vectors: Documented sustained safety and expression for 12-15 years in long-term follow-up [74]. |
Targeting Efficiency and Specificity
Achieving high expression in the target tissue while minimizing off-target effects is a major focus of platform engineering.
Table 3: Targeting Capabilities and Experimental Outcomes
| Platform / Strategy | Target Tissue/Cell | Experimental Model | Key Efficacy Metrics |
|---|---|---|---|
| LNP: OS4T Formulation [75] | Brain (Neurons, astrocytes, microglia) | Orthotopic glioblastoma mouse model | > 50-fold increase in mRNA translation in brain tissues vs. FDA-approved MC3 LNPs [75]. |
| LNP: ApoE-mediated Uptake [76] | Liver (Hepatocytes) | Multiple preclinical models | Natural tropism via ApoE association and LDL receptor binding on hepatocytes. Foundation of approved siRNA (Patisiran) and mRNA therapies [76]. |
| Engineered AAV (FUS-BBBO) [33] | Brain (Specific regions via FUS) | Mouse model (MRI-guided FUS) | >10-fold improvement in targeting specificity; enhanced neuronal transduction with reduced peripheral organ transduction [33]. |
| AAV (Natural Tropism) [74] | Liver | Human clinical trials for Hemophilia B | Consistent, long-term FIX expression at therapeutic levels (~40% of normal) enabling discontinuation of prophylaxis [74]. |
Experimental Protocols for Key Studies
Protocol: Evaluating Brain-Targeted LNP Delivery
This protocol is adapted from the study that developed the OS4T LNP for systemic mRNA delivery to the brain [75].
- LNP Formulation: Synthesize ionizable lipids derived from SR-57227, a 5-HT3 receptor ligand. Formulate LNPs using a microfluidic device by mixing an ethanol phase containing the ionizable lipid (OS4), DOPE, cholesterol, and DMG-PEG2k at a molar ratio of 40:40:60:0.75 with an aqueous phase containing the mRNA of interest (e.g., FLuc mRNA for benchmarking) in citrate buffer (pH 4.0).
- Post-modification with CPP: Conjugate the cell-penetrating peptide (CPP) Tat to the surface of the pre-formed OS4 LNPs via a Michael addition reaction to create the final OS4T LNP. Purify and characterize the size, PDI, and zeta potential.
- In Vivo Administration and Analysis:
- Systemic Injection: Administer OS4T LNPs intravenously to mice (e.g., at an mRNA dose of 0.6 mg/kg) via the tail vein. Include control groups (e.g., PBS, standard MC3 LNPs).
- Biodistribution Imaging: At a predetermined time point (e.g., 6 hours post-injection), image anesthetized mice using an in vivo imaging system (IVIS) to quantify luminescence signal from the brain and other major organs.
- Tissue Analysis: Euthanize the animals, harvest and homogenize brain tissues. Perform further analyses such as ELISA to quantify translated protein or immunohistochemistry to identify transduced cell types (neurons, astrocytes, microglia).
- Therapeutic Efficacy Model: In an orthotopic glioblastoma mouse model, administer multiple intravenous doses of OS4T LNPs loaded with engineered interleukin-12 (eIL-12) mRNA. Monitor tumor growth via imaging and record overall survival rates.
Protocol: Engineering AAV Vectors for Acoustically Targeted Brain Delivery
This protocol outlines the high-throughput in vivo selection process for engineering AAV vectors optimized for focused ultrasound blood-brain barrier opening (FUS-BBBO) [33].
- Library Generation: Create a diverse library of AAV capsid mutants by inserting a random 7-amino-acid peptide into the VP1 capsid protein of a parental AAV9 backbone.
- In Vivo Selection Round 1:
- FUS-BBBO: Anesthetize hSyn-Cre transgenic mice and use MRI-guided focused ultrasound to transiently open the blood-brain barrier at 4 predefined sites in one brain hemisphere.
- Viral Library Delivery: Immediately after FUS, intravenously inject the entire AAV mutant library (e.g., 1.3 × 10^9 unique variants) at a high dose.
- Incubation and DNA Extraction: Allow 2 weeks for gene expression. Euthanize the mice, harvest the targeted brain hemisphere, and extract genomic DNA.
- Cre-dependent PCR: Use Cre-dependent PCR to selectively amplify the viral genomes that have successfully transduced Cre-expressing neurons, thereby enriching for neuron-specific capsids.
- In Vivo Selection Round 2 (Quantitative Evaluation):
- Re-package the down-selected capsid variants into a new library.
- Repeat the FUS-BBBO and IV injection process in a new cohort of mice.
- Harvest both the FUS-targeted and the non-targeted contralateral brain hemispheres separately.
- Perform Cre-dependent PCR and next-generation sequencing (NGS) on DNA from both hemispheres to identify capsid variants that are highly enriched in the targeted hemisphere relative to the non-targeted one.
- Validation: Select the top candidate sequences (e.g., the 5 most enriched) based on their targeting specificity. Re-synthesize and package them as individual AAV vectors. Validate their performance against the wild-type AAV9 in subsequent experiments, quantifying transduction efficiency and specificity in the target brain region and peripheral organs.
The workflow for this sophisticated AAV engineering process is visualized below.
The Scientist's Toolkit: Key Reagents and Materials
Table 4: Essential Research Reagents for Gene Delivery Studies
| Reagent / Material | Function / Application | Example Use-Case |
|---|---|---|
| Ionizable Lipid (e.g., SM-102, DLin-MC3-DMA, OS4) [75] [71] | Critical for mRNA encapsulation and endosomal escape; protonates in acidic endosomes. | Core component of LNP formulations for mRNA vaccines and therapeutics. |
| Adeno-Associated Virus (AAV) Serotypes (e.g., AAV8, AAV9) [72] [74] | Engineered viral capsids with defined tissue tropism (e.g., liver, CNS). | Preferred vector for long-term gene therapy in post-mitotic tissues (e.g., Hemophilia B). |
| Cell-Penetrating Peptides (CPPs, e.g., Tat) [75] | Enhance cellular uptake and traversal of biological barriers (e.g., BBB). | Conjugated to LNPs (OS4T) to improve brain delivery efficiency. |
| hSyn-Cre Transgenic Mice [33] | Cre recombinase expressed under neuron-specific promoter (hSyn). | In vivo selection of AAV variants that specifically transduce neurons. |
| Focused Ultrasound (FUS) with Microbubbles [33] | Enables transient, localized opening of the blood-brain barrier. | Non-invasive, site-specific delivery of AAVs or LNPs to the brain from systemic circulation. |
The choice between LNPs and viral vectors is not a matter of superiority, but of strategic alignment with therapeutic goals. LNPs are the platform of choice for applications requiring transient expression, such as vaccines or short-term protein replacement, where a favorable safety profile and the option for repeated dosing are paramount. Their synthetic nature allows for rapid design and scalable manufacturing. The ongoing development of novel ionizable lipids and surface functionalization strategies, like the OS4T platform, is progressively overcoming their historical limitation of liver dominance and poor extrahepatic delivery efficiency [75] [76].
Conversely, viral vectors, particularly AAVs, remain the gold standard for interventions demanding durable, long-term gene expression to correct monogenic disorders, as evidenced by the clinical success in hemophilia B [74]. Their principal challenges include pre-existing immunity, potential for immunogenic reactions, and limited cargo capacity. However, advanced engineering techniques, such as the capsid evolution for FUS-BBBO, are directly addressing these hurdles by creating vectors with enhanced targeting specificity and reduced off-target effects [33].
The future of gene delivery lies in the continued refinement of both platforms and the potential for hybrid approaches. By leveraging the deep and growing understanding of the cellular and biophysical barriers to delivery—such as the intricate endosomal escape pathways for LNPs [28]—researchers can rationally design next-generation vectors. This progress will increasingly enable the precise control of therapeutic gene expression, both in its duration and location, ultimately unlocking the full potential of gene therapy for a wider array of human diseases.
The field of gene therapy is increasingly focused on two powerful delivery systems: viral vectors and lipid nanoparticles (LNPs). Viral vectors, derived from modified viruses like adenovirus (AdV) or adeno-associated virus (AAV), offer high delivery efficiency and, for some types, long-term gene expression through genome integration [1]. However, they face challenges such as pre-existing immune responses, potential insertional mutagenesis, and limited cargo capacity [3] [1]. In contrast, LNPs represent a versatile non-viral delivery platform. They are particularly valued for their lower immunogenicity, which allows for repeated dosing, a superior safety profile, and scalable production, as demonstrated by their pivotal role in mRNA COVID-19 vaccines [3] [77]. A typical LNP is a multi-component system where each lipid type plays a critical role. Ionizable lipids are the most crucial for complexing nucleic acids and facilitating endosomal escape, helper lipids (phospholipids and cholesterol) provide structural integrity and promote membrane fusion, and PEGylated lipids shield the particle to enhance stability and circulation time [78] [79]. This guide objectively compares the performance of these LNP components, providing supporting experimental data to outline their distinct roles and optimization strategies.
Component Roles and Experimental Optimization
Ionizable Lipids: The Engine of Delivery Efficiency
Ionizable lipids are the cornerstone of LNP performance, responsible for mRNA encapsulation, cellular uptake, and endosomal escape [80] [79]. Their charge changes with pH: neutral in the bloodstream to reduce toxicity, and positively charged in acidic endosomes to promote membrane disruption and cargo release [79].
Experimental Data and Performance Comparison A 2025 study directly compared four ionizable lipids—SM-102, ALC-0315, MC3, and 113-O12B—in mRNA-LNPs, revealing significant differences in their pharmacokinetics and biodistribution following intravenous (IV) and subcutaneous (SC) administration in mice [80] [81]. The table below summarizes the key quantitative findings.
Table 1: Performance Comparison of Ionizable Lipids in mRNA-LNP Formulations
| Ionizable Lipid | Key Pharmacokinetic Findings | Biodistribution of Expressed Protein | Notable Characteristics |
|---|---|---|---|
| SM-102 | Superior mRNA protection in plasma; highest bioavailability (approx. 3x higher than others after SC injection) [80]. | Comparable overall expression to ALC-0315; high early bioavailability drives expression [80]. | Used in Moderna's COVID-19 vaccine mRNA-1273 [79]. |
| ALC-0315 | Prolonged lipid exposure but reduced mRNA plasma concentration vs. SM-102 [80]. | Comparable overall expression to SM-102 [80]. | Used in Pfizer-BioNTech's COVID-19 vaccine BNT162b2 [79]. |
| MC3 | Longest terminal half-life; delayed mRNA expression [80]. | Liver-dominated after IV; local expression in skin and lymph nodes after SC [80]. | First approved for siRNA drug Onpattro (patisiran) [79]. |
| 113-O12B | Data included in study but not highlighted in abstract [80]. | Data included in study but not highlighted in abstract [80]. | -- |
The apparent pKa of the ionizable lipid at the LNP surface is a critical design parameter. The most effective pKa ranges are 6.2–6.5 for hepatic siRNA delivery and 6.6–6.9 for intramuscular mRNA vaccines [79]. The chemical structure—head group, linker (e.g., ester, ether), and tail chain length—directly influences the pKa, biodegradability, and efficacy of the lipid [79]. Esters, as used in SM-102 and ALC-0315, are often preferred for their biodegradability and safety profile [79].
PEGylated Lipids: The Stability and Targeting Modulator
PEGylated lipids, though typically constituting a small molar percentage (1.5-5%) of the formulation, are critical for LNP performance. They form a hydrophilic "protective barrier" on the LNP surface, which enhances colloidal stability, prevents aggregation, reduces serum protein adsorption, and prolongs systemic circulation [78] [79]. However, this benefit introduces the "PEG dilemma," where the same steric hindrance that provides stability can also impair cellular uptake and endosomal escape [78].
Experimental Data on PEG-Lipid Content Optimization A 2025 study systematically investigated the impact of varying DMG-PEG2000 content on LNP performance in vitro and in vivo [78]. The researchers formulated LNPs using a self-synthesized ionizable lipid and different molar ratios of DMG-PEG2000, loaded them with mRNA, and evaluated their properties and performance.
Table 2: Impact of DMG-PEG2000 Content on LNP Performance (In Vitro vs. In Vivo)
| DMG-PEG2000 Molar Ratio | Average Particle Size (nm) | Polydispersity Index (PDI) | Encapsulation Efficiency (%) | In Vitro Transfection (HeLa/DC2.4) | In Vivo Transgene Expression |
|---|---|---|---|---|---|
| 1.5% | ~80-100 (est.) | Low (est.) | >90% (est.) | Optimal | Moderate |
| 5% | ~80-100 (est.) | Low (est.) | >90% (est.) | Sub-optimal | Highest |
The study revealed a bell-shaped relationship between PEG content and efficacy [78]. A lower PEG content (1.5%) facilitated better cellular internalization in vitro, resulting in optimal transfection. In contrast, a higher PEG content (5%) was optimal in vivo, as it provided the stability needed for prolonged circulation and reaching target tissues [78]. This highlights a critical trade-off that must be managed during formulation.
Beyond content, the structure of the PEG-lipid matters. DMG-PEG2000, with its C14 lipid tail, is considered a "short-chain" PEG-lipid that can dissociate from the LNP after administration, enhancing cellular uptake and mRNA release [78]. Alternatives like DSPE-PEG have longer tail chains that provide more stable anchoring, which prolongs circulation but may also increase the potential for anti-PEG immune responses [78]. Recent innovations focus on PEG alternatives, such as polysarcosine-based shielding lipids, to address PEG-related immunogenicity concerns [82].
Helper Lipids: Cholesterol and Phospholipids
Helper lipids, comprising structurally supportive phospholipids and cholesterol, are essential for the integrity and function of LNPs.
- Phospholipids (e.g., DSPC, DOPE): These molecules form a lipid bilayer that stabilizes the LNP structure. Their selection significantly impacts delivery efficiency. Saturated phospholipids like DSPC are often used in commercial LNP formulations (at ~10 mol%) and are suitable for siRNA delivery [79]. Unsaturated phospholipids like DOPE, which promote membrane fusion, are often more effective for mRNA delivery, and replacing DSPC with DOPE has been shown to improve mRNA transfection efficiency in many studies [79].
- Cholesterol: This molecule is integrated into the lipid bilayer to reinforce membrane integrity and fluidity. It provides stability and helps prevent leakage of the encapsulated nucleic acid payload [78] [79].
Essential Experimental Protocols for LNP Development
LNP Formulation via Microfluidic Nanoprecipitation
The current standard method for preparing LNPs is microfluidic nanoprecipitation, which ensures reproducible and controlled formation of nanoparticles [77] [79].
Detailed Methodology [78]:
- Preparation of Lipid Stock Solutions: Dissolve the ionizable lipid, cholesterol, phospholipid (e.g., DOPE or DSPC), and PEG-lipid in anhydrous ethanol at precise concentrations (e.g., 100, 25, 25, and 50 mg/mL, respectively).
- Mixing Organic Phase: Combine the lipid stock solutions at a predetermined molar ratio (e.g., ionizable lipid : cholesterol : PEG-lipid : phospholipid = 40 : (50-X) : X : 10, where X is the variable PEG-lipid percentage).
- Rapid Mixing: Using a microfluidic device, rapidly mix the organic phase with an acidic aqueous buffer (e.g., 200 mM acetate buffer, pH 5.4) under continuous vortexing. The acidic pH protonates the ionizable lipid, enabling efficient complexation and encapsulation of the mRNA.
- Dialysis and Storage: Dialyze the resulting LNP suspension against a buffer such as PBS to remove ethanol and adjust the pH to physiological conditions. The final formulation can be stored at 4°C.
Key Characterization Assays
- Physicochemical Properties: Use Dynamic Light Scattering (DLS) to measure the hydrodynamic diameter, polydispersity index (PDI) indicating size distribution, and zeta potential (surface charge) of the LNPs [78]. Transmission Electron Microscopy (TEM) is used to visualize the morphology and structure of the particles after negative staining [78].
- Encapsulation Efficiency: This is quantified using a fluorescence-based assay like the Quant-iT RiboGreen RNA Assay [78]. Fluorescence is measured from samples with and without a detergent that disrupts the LNPs; the difference indicates the amount of RNA successfully encapsulated and protected from the dye.
- In Vitro Transfection Efficiency: Incubate LNPs with relevant cell lines (e.g., HeLa, DC2.4). Efficiency can be quantified by measuring the expression of a reporter protein (e.g., luciferase for luminescence, green fluorescent protein (GFP) for fluorescence) using a plate reader or flow cytometry [78].
- In Vivo Biodistribution and Efficacy: Administer LNPs to animal models (e.g., mice) via relevant routes (IV or SC). Analyze tissues over time for reporter protein expression (e.g., luciferase for bioluminescent imaging) or directly quantify mRNA and lipid concentrations in tissues and plasma to understand pharmacokinetics [80].
Visualization of LNP Workflow and Composition
The following diagram illustrates the typical workflow for developing and optimizing a Lipid Nanoparticle formulation, from component selection to final characterization.
Diagram 1: LNP Formulation Development Workflow.
The core structure of a functional LNP is built from four key lipid components, each playing a distinct role.
Diagram 2: Core LNP Components and Their Primary Functions.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential materials and technologies used in modern LNP research, as evidenced by recent studies and commercial developments.
Table 3: Essential Research Reagent Solutions for LNP Development
| Reagent / Technology | Function / Application | Key Features / Rationale |
|---|---|---|
| Ionizable Lipids (SM-102, ALC-0315) | Core component for mRNA delivery [80] [79]. | Clinically validated; biodegradable ester linkers; structure determines pKa and efficiency [79]. |
| PEG-Lipids (DMG-PEG2000) | Stability and pharmacokinetics modulator [78]. | Short C14 tail allows for dissociation, enhancing cellular uptake; industry standard [78]. |
| Helper Lipids (DSPC, DOPE) | Structural support and membrane fusion [79]. | DSPC provides stability; DOPE promotes fusogenicity, often enhancing mRNA delivery [79]. |
| PEG-Alternatives (Polysarcosine) | Biocompatible shielding to replace PEG [82]. | Aims to reduce PEG-related immunogenicity while maintaining stealth properties [82]. |
| Microfluidic Instruments (NanoAssemblr) | Scalable LNP formulation [77]. | Enables reproducible, controlled nanoprecipitation with high encapsulation efficiency (>90%) [77]. |
| Functionalized PEG-Lipids | Active targeting of specific tissues [83] [79]. | PEG chain can be conjugated with antibodies, peptides, or other ligands for targeted delivery [83]. |
| AI/ML Design Platforms | Accelerated LNP formulation discovery [77] [82]. | Uses machine learning to predict structure-function relationships, drastically reducing development time [77]. |
Optimizing LNP formulations is a complex, multi-parameter exercise that requires carefully balancing the roles of ionizable lipids, PEGylated lipids, and helper lipids. As the data shows, the choice of ionizable lipid (e.g., SM-102 vs. ALC-0315) directly impacts pharmacokinetics and biodistribution, while the PEG-lipid content requires a strategic trade-off between in vitro transfection and in vivo stability. The ongoing development of novel ionizable lipids, PEG alternatives, and AI-driven design platforms is poised to overcome current limitations in manufacturing, targeting, and immunogenicity. As these innovations mature, LNP technology is set to expand its impact far beyond vaccines and liver-targeted therapies, solidifying its role as a cornerstone of next-generation gene therapeutics and providing a powerful non-viral alternative in the gene delivery landscape.
The success of gene therapy hinges on the efficient and safe delivery of genetic material to target cells. For decades, viral vectors have been the dominant platform, prized for their high transduction efficiency and, in some cases, long-lasting gene expression. However, the clinical emergence of lipid nanoparticles (LNPs), particularly during the COVID-19 pandemic, has established a powerful non-viral alternative [3] [84]. This guide provides a objective comparison of these two platforms, focusing on the cutting-edge engineering strategies that are shaping their future: capsid engineering for viral vectors and targeted LNP design for non-viral systems.
The choice between viral and non-viral vectors is not a simple determination of superiority but a strategic decision based on the therapeutic goal. Viral vectors, particularly adeno-associated viruses (AAVs) and lentiviruses (LVs), are often selected for treatments requiring sustained, long-term gene expression, such as in monogenic disorders [3] [12]. In contrast, LNPs offer distinct advantages for applications requiring rapid, high-level protein production, reduced immunogenicity for repeat dosing, and greater payload flexibility, making them ideal for vaccines, transient gene editing, and cancer immunotherapy [3] [14] [84]. The following sections will dissect their performance, supported by experimental data and detailed methodologies driving the field forward.
Vector Performance: A Quantitative Comparison
The functional capabilities of viral vectors and LNPs differ significantly across key performance parameters. The tables below summarize these differences based on current technologies and approved therapies.
Table 1: Comparative Performance of Viral Vectors and LNPs
| Parameter | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Lipid Nanoparticles (LNPs) |
|---|---|---|---|
| Payload Capacity | ~4.7 kb [12] | ~8 kb [12] | >10 kb (DNA), highly flexible for RNA [14] [13] |
| Expression Duration | Long-term (years) [3] | Long-term (persistent due to integration) [3] | Transient (days to weeks for mRNA); months for DNA-LNPs [3] [14] |
| Immunogenicity | Moderate to High; pre-existing immunity common, limits re-dosing [3] [12] | Moderate [12] | Low immunogenicity; more suitable for repeated administration [3] [14] |
| Primary Applications | In vivo gene therapy for retinal diseases, SMA, hemophilia [12] | Ex vivo cell therapy (e.g., CAR-T, hematopoietic stem cells) [12] | mRNA vaccines, in vivo gene editing/silencing, protein replacement [3] [84] |
| Key Safety Concerns | Immune responses, hepatotoxicity at high doses [12] | Insertional mutagenesis [3] | Reactogenicity, lipid component toxicity [16] |
Table 2: Manufacturing and Commercial Considerations
| Consideration | Viral Vectors | LNPs |
|---|---|---|
| Scalability | Complex and costly manufacturing; requires packaging cell lines [3] [13] | Highly scalable; demonstrated by global mRNA vaccine production [3] [13] |
| Tropism/Targeting | Native tropism; can be re-targeted via capsid engineering [12] [85] | Primarily hepatic after IV injection; can be re-targeted via surface ligand conjugation and formulation tuning [16] [84] |
| Approved Therapies | 29 approved viral-based therapies (e.g., Luxturna, Zolgensma, CAR-T products) [12] | 6 approved non-viral based therapies (e.g., Onpattro for siRNA, mRNA COVID-19 vaccines) [12] |
Engineering Next-Generation Viral Vectors: Capsid Engineering
The capsid, the outer protein shell of a virus, is the primary determinant of its tropism, immunogenicity, and transduction efficiency. Engineering the capsid is therefore a major focus for improving viral vector performance.
Experimental Protocol: In Vivo Selection of Novel AAV Capsids
A common method for discovering novel AAV capsids with improved properties is Directed Evolution, which involves iterative selection rounds in vivo [12] [85].
- Library Creation: A diverse library of AAV capsid variants is generated via random peptide insertion, DNA shuffling, or error-prone PCR. Library size can exceed 10^6 unique variants.
- Library Administration: The pooled capsid library is administered to an animal model (e.g., mouse or non-human primate) via the intended route (e.g., intravenous, intramuscular).
- Recovery and Sequencing: After a set period, target tissues (e.g., liver, brain, retina) are harvested. Genomic DNA is isolated, and the AAV capsid DNA sequences are recovered via PCR and sequenced using next-generation sequencing (NGS).
- Data Analysis: Bioinformatic analysis of NGS data identifies capsid variants that are enriched in the target tissue relative to the input library and control tissues. This enrichment indicates successful targeting and transduction.
- Validation: Individual enriched capsid variants are packaged and re-administered to new animals to confirm their enhanced tropism and efficacy in a targeted gene delivery experiment.
Key Strategies in Capsid Engineering
- Capsid Modification for Immune Evasion: Researchers engineer mutations into the capsid surface to mask or eliminate immunogenic epitopes. This involves conjugating polymers or peptides to shield the capsid from neutralizing antibodies, prolonging its circulation time and enhancing transduction [85].
- Self-Inactivating (SIN) Vectors: Primarily used for lentiviral vectors, this strategy involves deleting enhancer/promoter sequences in the viral long terminal repeat (LTR) region. This modification reduces the risk of insertional mutagenesis by minimizing the potential for aberrant activation of nearby host genes after integration, thereby improving safety [85].
Engineering Next-Generation LNPs: Targeted Delivery and AI-Driven Design
While first-generation LNPs excel in hepatic delivery and intramuscular vaccination, a key research frontier is achieving targeted extrahepatic delivery. This involves both passive targeting through formulation and active targeting using surface ligands.
Experimental Protocol: Screening Ionizable Lipids for Enhanced Potency
A recent study demonstrated a high-throughput approach to develop highly potent LNPs for mRNA vaccines [86].
- Rational Lipid Design and Library Synthesis: A library of novel ionizable lipids was designed based on chemical features known to enhance delivery, such as cyclic structures for improved efficiency and ester groups for enhanced biodegradability [86].
- High-Throughput Formulation and Screening: LNPs encapsulating mRNA encoding a reporter gene (e.g., luciferase) were formulated using microfluidics. This library of LNP formulations was then screened in vivo in mice. The bioluminescent signal was measured to quantify protein expression and identify the top-performing LNP candidate [86].
- Iterative Optimization: The lead LNP structure (AMG1541) was used as a template to create a second-generation library of variants, which underwent another round of in vivo screening to finalize the optimal candidate [86].
- Efficacy and Mechanism Evaluation: The final LNP was used to deliver an mRNA flu vaccine. The immune response (antibody titers) was compared to an FDA-approved LNP (SM-102) at various doses. Further mechanistic studies assessed endosomal escape efficiency, uptake by antigen-presenting cells, and biodistribution to lymph nodes [86].
- Key Findings: The novel LNP (AMG1541) generated a comparable antibody response to the SM-102 LNP at a 100-fold lower mRNA dose. This was attributed to more efficient endosomal escape and greater accumulation in lymph nodes, highlighting the impact of ionizable lipid structure on potency [86].
Advanced Strategies in LNP Engineering
- AI-Driven LNP Formulation: Machine learning (ML) models are trained on large datasets of LNP compositions and their in vivo performance outcomes. For instance, graph neural networks can predict RNA-LNP binding affinity, while generative adversarial networks (GANs) can design novel ionizable lipid structures with desired pKa and branching patterns, dramatically accelerating the development timeline [16].
- Overcoming DNA-LNP Toxicity: A breakthrough in DNA-LNP development involved identifying the cGAS-STING inflammatory pathway as the cause of lethal immune reactions in earlier attempts. Researchers mitigated this by incorporating a natural anti-inflammatory molecule, nitro-oleic acid (NOA), into the DNA-carrying particles. This intervention eliminated fatal reactions in mice and enabled long-term (∼6 months) transgene expression from a single dose, paving the way for DNA-based gene therapies using LNPs [14].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and technologies essential for research in capsid engineering and advanced LNP development.
Table 3: Key Research Reagents and Solutions
| Research Reagent / Technology | Function in Research |
|---|---|
| Ionizable Lipids (e.g., SM-102, AMG1541) | The key functional component of LNPs; critical for encapsulating nucleic acids, facilitating endosomal escape, and determining efficiency and tropism [86]. |
| Capsid Library | A diverse pool of AAV capsid variants (created via peptide display, mutagenesis) used in directed evolution experiments to select for clones with desired tropism [12] [85]. |
| Microfluidic Mixers | Instruments (e.g., nanoassemblers) used for the precise, reproducible, and scalable formulation of LNPs, ensuring consistent particle size and encapsulation efficiency [87]. |
| Targeting Ligands | Molecules (e.g., antibodies, peptides, sugars like GalNAc) conjugated to the surface of viral vectors or LNPs to actively target specific cell receptors and enhance tissue-specific delivery [16] [84] [85]. |
| Machine Learning Models | Software and algorithms (e.g., GNNs, GANs) used to virtually screen lipid libraries, predict LNP properties, and generate novel lipid structures, reducing reliance on purely experimental HTS [16] [88]. |
Visualizing Experimental Workflows
The following diagrams illustrate the core experimental workflows for developing next-generation vectors.
Directed Evolution Workflow for AAV Capsids
AI-Driven LNP Screening Workflow
LNP-Mediated mRNA Delivery Mechanism
The evolution of gene delivery is being propelled by sophisticated engineering of both viral and non-viral vectors. Capsid engineering endeavors to refine viral vectors, enhancing their safety and precision, while research in targeted LNPs and AI-driven design seeks to expand the utility of non-viral platforms beyond their current limitations. The choice between these platforms remains context-dependent, dictated by the specific therapeutic application, desired duration of expression, and safety considerations. The parallel advancements in both fields, coupled with an emerging understanding that they may be complementary, promise to unlock a new era of gene therapies for a broader range of human diseases. Future directions will likely see increased integration of computational and AI tools, such as quantum machine learning for stability prediction, to further accelerate the rational design of next-generation vectors [16] [88].
Head-to-Head Analysis: Safety, Efficacy, and Commercial Viability
The selection of a delivery vector is a critical determinant in the safety and efficacy of any gene therapy. Within this landscape, lipid nanoparticles (LNPs) and viral vectors represent two leading technologies with distinctly different safety profiles. For researchers and drug development professionals, understanding the nuances of immunogenicity, risk of insertional mutagenesis, and overall toxicity is paramount for rational vector selection and therapeutic design. Viral vectors, derived from naturally evolved viruses, are engineered for safety but retain certain inherent biological characteristics that can trigger immune responses or pose genotoxic risks [52] [89]. In contrast, LNPs are synthetically designed delivery systems that offer a different set of advantages and limitations, primarily centered on their biocompatibility and transient expression profile [20] [73]. This guide provides a detailed, evidence-based comparison of these platforms, framing their safety attributes within the context of modern gene therapy development.
Comparative Safety Profiles at a Glance
The table below summarizes the key safety characteristics of LNPs and viral vectors, providing a high-level overview for initial assessment.
Table 1: Head-to-Head Comparison of Key Safety Profiles
| Safety Parameter | Lipid Nanoparticles (LNPs) | Viral Vectors (Adeno-Associated Viruses) | Viral Vectors (Lentiviruses) |
|---|---|---|---|
| Immunogenicity | Low to moderate; allows for repeated dosing [3] [11]. | High; pre-existing immunity is common, and therapy can trigger strong inflammatory responses, limiting re-dosing [3] [89] [11]. | Moderate to high; can trigger immune responses [89]. |
| Risk of Insertional Mutagenesis | None; mRNA operates in the cytoplasm and does not enter the nucleus, eliminating the risk of insertional mutagenesis [90] [91]. | Very low; predominantly remain episomal, though rare integration events can occur [11]. | Yes; integrate into the host genome, posing a recognized risk of insertional mutagenesis that can lead to genotoxicity and cancer [3] [89]. |
| Primary Toxicity Concerns | Acute inflammatory reactions; component-related toxicity (e.g., ionizable lipids, PEG) [20] [91]. | Dose-dependent liver toxicity (acute liver failure reported); immune-mediated toxicities [11]. | Insertional mutagenesis; immune responses to viral components [89]. |
| Duration of Transgene Expression | Transient (days to weeks), suitable for short-term protein production [3]. | Long-term (potentially years) and stable expression from episomal DNA [3] [11]. | Long-term or permanent due to genomic integration [3]. |
Deep Dive into Immunogenicity
Immunogenicity remains one of the most significant hurdles in gene therapy, as it can reduce efficacy, cause adverse events, and prevent re-dosing.
Immunogenicity of Viral Vectors
Viral vectors can stimulate both innate and adaptive immune responses. The host's immune system recognizes the viral capsid or envelope proteins, leading to vector neutralization and inflammatory reactions.
- Pre-existing Immunity: A major challenge for AAV and adenoviral vectors is the high prevalence of pre-existing neutralizing antibodies in the human population due to prior natural infections [89] [11]. These antibodies can bind to the vector and prevent it from reaching the target cells, rendering the therapy ineffective.
- Cellular Immune Responses: The viral capsid can be processed and presented by antigen-presenting cells, leading to cytotoxic T lymphocyte (CTL) responses that eliminate transduced cells and abrogate long-term transgene expression [89]. This is a key factor in the decline of therapeutic effect observed in some clinical trials.
- Clinical Consequences: The immune response against viral vectors not only limits efficacy but also poses a significant safety risk. In severe cases, such as the historic 1999 trial using an adenoviral vector, an overwhelming systemic inflammatory response led to patient death [11]. Furthermore, these immune responses make it exceedingly difficult to re-administer the therapy, as seen in cases like that of a patient treated in 1999 for a neuromuscular disorder, who can no longer receive viral vector therapies due to the risk of a dangerous cytokine storm [11].
Immunogenicity of Lipid Nanoparticles
LNPs are generally less immunogenic than their viral counterparts, but they are not inert.
- Component-Driven Inflammation: The ionizable lipid component is a primary trigger for innate immune responses, which can lead to transient inflammatory reactions such as fever and fatigue [20]. These are often manageable and short-lived.
- Anti-PEG Immunity: Polyethylene glycol (PEG)-lipids, used to stabilize LNPs, can induce anti-PEG antibodies, which may accelerate blood clearance of the LNPs and, in rare cases, contribute to allergic reactions [20] [91].
- Advantage for Re-dosing: The lower and more manageable immunogenicity of LNPs is a key advantage for therapies requiring multiple administrations. Unlike viral vectors, LNPs do not typically induce a memory immune response against the delivery vehicle itself, making repeated dosing feasible [3] [11].
Table 2: Experimental Assays for Profiling Immunogenicity
| Assay Type | Target of Analysis | LNP Workflow | Viral Vector Workflow |
|---|---|---|---|
| In Vitro Assays | Innate immune activation | Incubate LNP with human peripheral blood mononuclear cells (PBMCs) or reporter cell lines; measure cytokine (e.g., IFN-γ, IL-6) production via ELISA [20]. | Incubate vector with PBMCs; assess cytokine release and T-cell activation via flow cytometry [89]. |
| In Vivo Models | Integrated immune response | Administer LNP to animal models (e.g., mice); monitor for acute inflammatory signs and quantify plasma cytokine levels [20] [92]. | Administer vector to animal models; evaluate vector neutralization, T-cell infiltration in tissues, and loss of transgene expression over time [89]. |
| Human Seroprevalence | Pre-existing immunity | Not typically required. | Screen human serum samples for neutralizing antibodies (NAbs) against the viral capsid using cell-based transduction inhibition assays [52] [11]. |
Deep Dive into Insertional Mutagenesis
Insertional mutagenesis refers to the disruption of a host gene by the integration of foreign DNA, which can potentially lead to the activation of oncogenes or inactivation of tumor suppressor genes.
Risk Profile of Viral Vectors
The risk is highly dependent on the viral vector platform and its biological life cycle.
- Lentiviral Vectors: These are integrating vectors designed to insert their genetic payload into the host genome to achieve long-term expression in dividing cells. This process carries an inherent risk of insertional mutagenesis [3] [89]. While self-inactivating (SIN) designs have improved safety, the risk of genotoxicity leading to clonal expansion and cancer remains a key safety consideration in clinical trial design and long-term patient monitoring.
- Adeno-Associated Virus (AAV) Vectors: AAV vectors were long believed to exist almost exclusively as non-integrating episomes. However, advanced sequencing techniques have shown that they can integrate into the host genome at low frequencies, particularly in regions of DNA damage [11]. The clinical significance of this is still under investigation, but it is considered a much lower risk compared to lentiviruses.
- Adenoviral Vectors: These vectors remain episomal and do not integrate into the host genome, thereby presenting a negligible risk of insertional mutagenesis [89].
Innate Safety of LNPs for mRNA Delivery
A fundamental safety advantage of LNP-mediated mRNA delivery is the complete absence of insertional mutagenesis risk. The mRNA delivered by LNPs is transient and functions entirely in the cytoplasm; it does not need to enter the nucleus and has no mechanism for integration into the host genome [90] [91]. This makes mRNA-LNPs an exceptionally safe platform from a genotoxicity standpoint. For DNA-based therapies, non-viral delivery systems like LNPs carrying plasmid DNA also largely remain episomal, though the theoretical risk of random integration exists at a vastly lower frequency than with retroviral vectors.
Deep Dive into Toxicity
Beyond immunogenicity and genotoxicity, other acute and organ-specific toxicities are critical to evaluate.
Toxicity of Viral Vectors
- Dose-Limiting Liver Toxicity: This is a particularly serious concern for systemically administered AAV vectors. The liver acts as a primary sink for AAVs, leading to high viral uptake in hepatocytes [11]. This, combined with the host's innate and adaptive immune response to the capsid, can lead to acute liver injury and, in severe cases, liver failure, as reported in several clinical trials for neuromuscular diseases [11].
- Toxicity from DNA Contaminants: A manufacturing-related safety concern for AAVs involves the potential packaging of plasmid backbone sequences from the bacterial systems used to produce the vectors. These bacterial DNA sequences are potentially toxic if expressed in human cells and have been linked to neurotoxicity in animal models [93]. Novel manufacturing approaches using "gutless" plasmids with safe human DNA backbones are being developed to mitigate this risk [93].
Toxicity of Lipid Nanoparticles
- Component-Specific Toxicity: The toxicity profile of LNPs is directly linked to the chemical structure of their lipid components. First-generation cationic lipids were often associated with significant cytotoxicity [20]. Modern, optimized ionizable lipids have greatly improved the safety profile, but the potential for off-target effects and organ-specific accumulation remains an active area of research and optimization [20] [91].
- Infusion-Related Reactions: A common, though often manageable, toxicity associated with LNP administration is the occurrence of acute infusion-related reactions, which can include symptoms like fever, chills, and headache, and are believed to be driven by innate immune activation and cytokine release [90].
Essential Experimental Protocols for Safety Profiling
Robust preclinical safety assessment is non-negotiable for clinical translation. Below are detailed methodologies for key experiments.
Protocol for In Vivo Immunogenicity Assessment
Objective: To evaluate the potential of a gene delivery vector to elicit innate and adaptive immune responses in a live animal model.
Materials:
- Test articles: LNP formulation or viral vector.
- Control: Buffer or empty vehicle.
- Animal model: C57BL/6 mice (6-8 weeks old).
- Equipment: Flow cytometer, ELISA plate reader, microcentrifuge.
- Reagents: ELISA kits for cytokines (IFN-α, IFN-γ, IL-6, TNF-α), flow cytometry antibodies (CD4, CD8, CD44, CD62L), plasma separation tubes.
Procedure:
- Dosing and Sample Collection: Randomize mice into groups (n=5-10) and administer a single intravenous dose of the vector or control. Collect blood via retro-orbital bleeding at predetermined time points (e.g., 2, 6, 24 hours post-dose for innate response; 7, 14, 28 days for adaptive response). Centrifuge blood to collect plasma and store at -80°C.
- Cytokine Analysis (ELISA): Thaw plasma samples on ice. Follow manufacturer's instructions for the cytokine ELISA kits. Briefly, add samples and standards to pre-coated plates, incubate, wash, add detection antibody, incubate, wash, add substrate, and measure absorbance. Determine cytokine concentrations from the standard curve [20] [89].
- T-Cell Activation Analysis (Flow Cytometry): At terminal time points (e.g., day 14), isolate splenocytes. Stimulate cells ex vivo with PMA/ionomycin in the presence of a protein transport inhibitor. Stain cells with surface markers (CD4, CD8), then fix, permeabilize, and stain for intracellular cytokines (IFN-γ, IL-2). Analyze using flow cytometry to quantify the percentage of activated, cytokine-producing T cells [89].
Protocol for Assessing Genotoxicity and Insertional Mutagenesis
Objective: To determine the potential for a vector to integrate into the host genome and disrupt gene function.
Materials:
- Target cells: HeLa cells or primary human hematopoietic stem cells (HSCs).
- Test vectors: Lentiviral vector (positive control), AAV vector, LNP-mRNA.
- Reagents: Cell culture media, DNA extraction kit, LAM-PCR or LIDE-PCR reagents, next-generation sequencing (NGS) library prep kit, bioanalyzer.
- Equipment: Thermal cycler, NGS sequencer, cell culture incubator.
Procedure:
- In Vitro Transduction/Transfection: Transduce/transfect cells at a high multiplicity of infection (MOI) or appropriate dose. Culture cells for approximately 14 days to allow for genomic integration and clonal expansion.
- Genomic DNA Extraction: Harvest cells and extract high-molecular-weight genomic DNA using a commercial kit. Quantify and assess DNA quality via spectrophotometry and gel electrophoresis.
- Integration Site Analysis (LAM-PCR): Digest genomic DNA with a frequent-cutting restriction enzyme (e.g., MseI). Perform linear amplification using biotinylated primers specific to the vector's LTR or ITR sequence. Capture the amplified single-stranded DNA on streptavidin-coated magnetic beads. Ligate a double-stranded linker cassette to the unknown genomic end of the fragments. Perform nested PCR amplification using primers for the linker and the vector. Purify the PCR products and prepare for NGS [89].
- Bioinformatic Analysis: Process NGS reads to map vector-genome junctions to the reference human genome. Identify the genomic location of integration sites and analyze for preferences near oncogenes or tumor suppressors (e.g., in transcription start sites).
Visualizing Key Safety Pathways and Workflows
The following diagrams illustrate the fundamental mechanisms underlying the primary safety concerns of both vector platforms.
Immunogenicity and Toxicity Pathways
Diagram 1: Immunogenicity and Toxicity Pathways. This diagram contrasts the immune pathways activated by viral vectors, which can lead to severe toxicity and preclude re-dosing, with those of LNPs, which are generally more manageable and permit re-dosing [3] [89] [11].
Genotoxicity Risk Workflow
Diagram 2: Genotoxicity Risk Workflow. This chart illustrates the fundamental difference in genotoxicity risk: Lentiviral vectors integrate into the host genome, posing a risk of insertional mutagenesis, while mRNA delivered by LNPs functions in the cytoplasm and presents no such risk [90] [91] [89].
The Scientist's Toolkit: Key Research Reagents and Materials
The table below lists essential tools and materials used in the profiling and development of safe gene delivery vectors.
Table 3: Essential Reagents for Safety Profiling Experiments
| Reagent / Material | Function in Research | Application Example |
|---|---|---|
| Ionizable Lipids | The key functional component of LNPs for encapsulating nucleic acids and enabling endosomal escape; its chemical structure dictates efficacy and inflammatory potential [20]. | Screening novel ionizable lipids in vitro to identify candidates with high delivery efficiency and low cytokine induction [20] [92]. |
| PEG-Lipids | A surface-active lipid used to stabilize LNP formulations and modulate pharmacokinetics; can be a source of immunogenicity [20] [91]. | Optimizing the molar ratio and type of PEG-lipid in a formulation to balance particle stability, circulation time, and immunogenic profile. |
| AAV Serotype Libraries | Different AAV capsids (serotypes) exhibit distinct tissue tropisms and immunogenic properties. | Screening various serotypes (e.g., AAV8, AAV9, AAVrh74) in vivo to identify the one with optimal targeting to the tissue of interest and lowest immunogenicity in a given model [11]. |
| Cytokine ELISA Kits | Quantitative measurement of specific cytokine proteins (e.g., IFN-γ, IL-6) in cell culture supernatant or animal plasma. | Profiling the innate immune response following LNP or viral vector administration in mice by measuring plasma cytokine levels at various time points [20] [89]. |
| LAM-PCR Reagents | A core set of enzymes and linkers for performing linear-amplification mediated PCR to identify genomic integration sites of viral vectors [89]. | Assessing the genomic integration profile and potential for insertional mutagenesis of a novel lentiviral vector in transduced hematopoietic stem cells. |
| Anti-PEG Antibodies | Reagents used to detect and quantify the presence of antibodies against PEG in serum samples. | Evaluating the immunogenicity of PEG-lipids in repeat-dose toxicity studies to determine if anti-PEG antibodies are generated and if they impact LNP pharmacokinetics [20]. |
In the field of gene therapy, the choice of delivery vector is paramount, directly influencing the efficiency, durability, and safety of the therapeutic intervention. The core thesis of lipid nanoparticles (LNPs) versus viral vectors extends to a fundamental comparison of their operational metrics: transfection efficiency (for non-viral LNPs) and transduction efficiency (for viral vectors), along with the resulting expression kinetics of the delivered genetic material. This guide provides an objective, data-driven comparison of these critical performance parameters to inform researchers, scientists, and drug development professionals in their selection and development of gene delivery systems.
Defining the Core Metrics
Transfection Efficiency of Lipid Nanoparticles
Transfection refers to the process of introducing nucleic acids into cells using non-viral methods. For Lipid Nanoparticles (LNPs), this involves the encapsulation of genetic material and its delivery into the cell cytoplasm via membrane fusion [3]. Efficiency is typically quantified as the percentage of cells that successfully express the delivered transgene. It is a highly variable metric dependent on LNP composition, cell type, and experimental conditions. Recent studies report that optimizing LNP composition, such as by incorporating a novel glutamate-cholesterol (GA–Chol) derivative, can improve in vitro transfection efficiency by approximately 10 to 20-fold in certain cell lines compared to standard formulations [94].
Transduction Efficiency of Viral Vectors
Transduction describes the delivery of genetic material into a cell using a viral vector. These vectors, such as lentiviruses (LVs) or adenoviruses (AVs), leverage the natural infectious mechanisms of viruses to enter cells [95]. Transduction efficiency is also measured as the percentage of cells expressing the transgene and is generally high. In clinical manufacturing of CAR-T cells, transduction efficiencies typically range from 30% to 70% [95]. This efficiency is influenced by factors including viral tropism, target cell type, and the multiplicity of infection (MOI).
Expression Kinetics
Expression kinetics describe the temporal profile of protein production from the delivered genetic material. This is a key differentiator between the two vector systems:
- LNPs are predominantly used for RNA delivery (e.g., mRNA), which leads to transient gene expression within the cytoplasm. This makes them ideal for vaccines or therapies requiring short-term protein production [3].
- Viral Vectors can be engineered for sustained expression. Certain viral vectors, like lentiviruses and gamma-retroviruses, integrate their genetic payload into the host genome, enabling long-term or even permanent gene expression, which is suitable for correcting genetic defects [3] [95].
Quantitative Comparison of Efficiency and Expression
The following tables synthesize key quantitative data from the literature to facilitate a direct comparison between LNP and viral vector systems.
Table 1: Key Performance Metrics for Gene Delivery Vectors
| Metric | Lipid Nanoparticles (LNPs) | Lentiviral Vectors (LVs) | Adeno-Associated Viruses (AAVs) | Gamma-Retroviruses (γRVs) |
|---|---|---|---|---|
| Reported Transfection/Transduction Efficiency | ~10-20 fold improvement with GA-Chol LNPs in vitro [94] | 30-70% in clinical CAR-T cell manufacturing [95] | High efficiency for specific cell types (e.g., DCs) [95] | Robust in ex vivo activated T cells [95] |
| Typical Multiplicity of Infection (MOI) | Not Applicable (N/A) - dose is lipid/nucleic acid ratio | Requires optimization; varies by cell type [95] | Requires optimization; varies by cell type [95] | Requires optimization; varies by cell type [95] |
| Vector Copy Number (VCN) | N/A - non-integrating | Generally kept below 5 copies/cell clinically [95] | N/A - non-integrating | Generally kept below 5 copies/cell clinically [95] |
| Kinetics of Expression | Transient (days) [3] | Long-term / Permanent (integration-dependent) [3] [95] | Prolonged (non-integrating, but episomal persistence) [95] | Long-term / Permanent (integration-dependent) [95] |
| Primary Expression Location | Cytoplasmic [3] | Nuclear / Genome-integrated [3] | Nuclear (episomal) [95] | Nuclear / Genome-integrated [95] |
Table 2: Comparative Analysis of Advantages and Limitations
| Characteristic | Lipid Nanoparticles (LNPs) | Viral Vectors (e.g., LV, AAV) |
|---|---|---|
| Immune Response | Lower immunogenicity; suitable for repeated dosing [3] | Can trigger significant immune responses; limits re-dosing [3] [95] |
| Scalability & Manufacturing | Relatively easy to scale up; commercially demonstrated [3] | More complex and costly large-scale production [3] |
| Tissue Targeting | Can be engineered, but capabilities are still advancing (e.g., GA-Chol LNPs for localized delivery) [3] [94] | High precision; can be engineered for specific tissue tropism (e.g., liver, muscle) [3] |
| Safety Profile | Favorable; low risk of insertional mutagenesis [3] | Risk of insertional mutagenesis (integrating vectors); immunogenicity [3] [95] |
| Payload Capacity | Versatile; can deliver mRNA, siRNA, CRISPR components [3] | Limited by viral capsid size (e.g., ~4.7 kb for AAV, ~8 kb for AV) [95] |
Experimental Protocols for Efficiency Measurement
Protocol: Measuring LNP Transfection Efficiency via Flow Cytometry
This protocol is adapted from studies optimizing LNP-mediated CAR-T cell engineering [96] and localized mRNA delivery [94].
- LNP Formulation: Prepare LNPs using microfluidic mixing. A standard composition includes an ionizable lipid (e.g., D-Lin-MC3-DMA), a helper phospholipid (e.g., DSPC), cholesterol or a derivative (e.g., GA-Chol), and a PEGylated lipid (e.g., DMG-PEG2k) in a molar ratio such as 50:10:38.5:1.5. Encapsulate a reporter mRNA, such as GFP mRNA.
- Cell Transfection: Culture target cells (e.g., activated primary human T cells [96] or HEK293T/HeLa cells [94]) in appropriate media. Transfect cells with the formulated LNPs. For T cells, consider supplementing the medium with proteins like ApoE to enhance binding to LDL receptors and improve transfection efficiency [96].
- Incubation and Analysis: Incubate cells for 24-48 hours to allow for protein expression. Harvest the cells and analyze using flow cytometry. The percentage of GFP-positive cells relative to the total cell population quantifies the transfection efficiency.
Protocol: Measuring Viral Transduction Efficiency via Flow Cytometry and VCN
This protocol is standard in immune cell therapy manufacturing, as reviewed in [95].
- Vector Production and Titration: Produce replication-deficient viral vectors (e.g., Lentivirus with a VSV-G envelope) and determine the viral titer (e.g., transducing units per mL).
- Cell Transduction: Pre-activate target cells (e.g., T cells using CD3/CD28 antibodies) to upregulate viral receptor expression. Transduce the cells at a specific Multiplicity of Infection (MOI) in the presence of transduction enhancers (e.g., polybrene). Spinoculation (centrifugation during transduction) can be employed to enhance cell-vector contact.
- Efficiency Assessment: After a suitable incubation period (e.g., 72 hours), assess transduction efficiency by flow cytometry for a surface or intracellular marker (e.g., the CAR transgene or GFP).
- Safety Assessment (Vector Copy Number): Genomic DNA is extracted from the transduced cell population. The average Vector Copy Number (VCN) is quantified using droplet digital PCR (ddPCR), with clinical programs generally maintaining VCN below 5 copies per cell [95].
Visualizing Intracellular Trafficking and Key Workflows
The differential pathways and kinetics of LNP and viral vector systems can be visualized through the following diagrams.
Diagram Title: Intracellular Trafficking Pathways for LNPs and Viral Vectors
Diagram Title: Core Workflow for Measuring Delivery Efficiency
The Scientist's Toolkit: Key Research Reagents
Successful experimentation in gene delivery requires a suite of specialized reagents and tools. The following table details essential solutions for working with LNPs and viral vectors.
Table 3: Essential Research Reagents for Gene Delivery Studies
| Reagent / Material | Function / Application | Example(s) / Notes |
|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs; condenses nucleic acids and facilitates endosomal escape [3] [94]. | D-Lin-MC3-DMA, C12-200 [96] [94]. |
| Structural Lipids | Stabilize LNP structure and contribute to bilayer integrity [3] [94]. | DSPC (phospholipid), Cholesterol or derivatives (e.g., GA-Chol) [94]. |
| PEGylated Lipids | Improve LNP stability, reduce aggregation, and modulate pharmacokinetics [3] [94]. | DMG-PEG2k [94]. |
| Viral Vector Systems | Engineered viruses for high-efficiency gene delivery. | Lentivirus (LV), Adenovirus (AV), Adeno-associated virus (AAV) [95]. |
| Transduction Enhancers | Chemicals or polymers that increase viral transduction efficiency [95]. | Polybrene, Retronectin. |
| Cell Culture Supplements | Support cell viability, activation, and post-transduction/transfection function. | IL-2 (for T cells), IL-15 (for NK cells) [95]. |
| Reporter Genes | Enable quantification of delivery efficiency and expression kinetics. | Green Fluorescent Protein (GFP), Firefly Luciferase (FLuc) [94] [97]. |
| Flow Cytometry Assays | Gold-standard for quantifying the percentage of successfully modified cells. | Used with reporter genes or antibody staining for surface CAR expression [96] [95]. |
| qPCR/ddPCR Assays | Quantify vector copy number (VCN) for integrating viral vectors to assess safety [95]. | Droplet digital PCR (ddPCR) is the gold standard for VCN [95]. |
The choice between lipid nanoparticles and viral vectors is not a matter of one being universally superior, but rather which is optimal for a specific therapeutic goal. LNPs excel in applications requiring transient expression and re-dosing, such as vaccines or some cancer immunotherapies, offering a favorable safety profile and scalable manufacturing. Viral vectors remain the cornerstone for therapies demanding high-efficiency delivery and long-term, stable gene expression, as in many monogenic disorders. The ongoing innovation in both fields—such as the development of novel lipids for localized LNP delivery [94] and the creation of safer, next-generation viral vectors [98]—continues to expand the possibilities for gene therapy. The decision must be guided by a careful evaluation of the efficiency metrics and expression kinetics detailed in this guide.
The advancement of gene therapies is intrinsically linked to the development of effective delivery vectors, with lipid nanoparticles (LNPs) and viral vectors representing the two foremost technologies. While their biological performance is often compared, their scalability and manufacturing economics are equally critical for determining their suitability for different therapeutic applications. The production pathway for each vector presents a distinct set of challenges and cost implications that directly influence clinical development timelines, commercial viability, and ultimate patient access [3]. This guide provides a detailed, objective comparison of the production complexity and cost for these platforms, synthesizing current market data and technical insights to inform strategic decision-making for researchers, scientists, and drug development professionals.
The global demand for both vector systems is experiencing significant growth, driven by an expanding pipeline of gene therapies, vaccines, and other advanced therapeutics. Quantitative market analysis reveals the scale and velocity of this expansion.
Table 1: Comparative Market Overview for LNP and Viral Vector Manufacturing
| Feature | Lipid Nanoparticle (LNP) Market | Viral Vector Manufacturing Market |
|---|---|---|
| Market Size (Base Year) | USD 1.16 Billion (estimated for 2025) [99] | US$ 1.40 Billion (estimated for 2025) [100] |
| Market Size (Forecast Year) | USD 2.88 Billion by 2032 [99] | US$ 3.75 Billion by 2032 [100] |
| Compound Annual Growth Rate (CAGR) | 13.9% (2025-2032) [99] | 15.11% (2025-2032) [100] |
| Projected U.S. Market Value | US$ 519.18 Billion by 2033 [101] | Information not specified in search results |
| Key Growth Drivers | RNA-based therapeutics, vaccines, targeted drug delivery, investments in biopharmaceutical R&D [99] [101] | Rising incidence of genetic disorders and cancers, approvals for gene therapies, success of viral vector-based vaccines [100] [102] |
Production Complexity and Cost Analysis
The manufacturing processes for LNPs and viral vectors differ fundamentally, leading to divergent profiles in terms of complexity, scalability, and cost structure.
Lipid Nanoparticle (LNP) Manufacturing
LNPs are synthetic, self-assembling particles. Their production, while technically advanced, benefits from a relatively streamlined process.
- Process Overview: LNP formulation typically involves rapid mixing of an aqueous phase containing the nucleic acid payload (e.g., mRNA) with an ethanol phase containing ionizable lipids, helper phospholipids, cholesterol, and PEG-lipids [87]. This process can be achieved with microfluidic devices or T-mixers, enabling precise control over particle size and encapsulation efficiency.
- Scalability: LNP production is considered highly scalable. Technologies like Precision NanoSystems' NxGen can generate LNP batches up to 400 liters, sufficient for both clinical studies and large-scale commercial production [103]. The relative ease of scaling up was demonstrated during the global rollout of COVID-19 mRNA vaccines [3].
- Cost and Challenges: A primary challenge is the high cost of specialized lipid excipients and the complexity of optimizing LNP formulations for specific tissue tropism [103] [99]. The manufacturing process itself forces drug developers to rely on contract development and manufacturing organizations (CDMOs), with more than 50 companies currently offering such services [103]. Regulatory uncertainty surrounding the long-term safety of nanomedicines also adds to the development burden [99].
Viral Vector Manufacturing
Viral vector production relies on complex biological systems, using engineered mammalian cells to produce replication-incompetent viruses.
- Process Overview: Manufacturing involves upstream processes (cell culture and transfection/infection to produce the virus) and downstream processes (purification and concentration). This can be done via transient transfection or using stable producer cell lines [100] [102].
- Scalability and Complexity: Large-scale production is inherently complex and resource-intensive. It requires advanced infrastructure, such as GMP-certified facilities and sophisticated bioreactors [100]. While suspension cell culture systems are improving scalability, the process faces challenges like low vector yields, downstream purification bottlenecks, and batch-to-batch variability [102]. The cost of producing a single patient-specific vector dose can reach tens of thousands of dollars [102].
- Cost Drivers: The high production costs are driven by the need for sophisticated facilities, extensive validation, and stringent quality control [102]. The field also faces a shortage of skilled professionals, which can slow operational expansion [100].
Table 2: Head-to-Head Comparison of Production Attributes
| Attribute | Lipid Nanoparticles (LNPs) | Viral Vectors |
|---|---|---|
| Production Process | Biochemical synthesis and self-assembly [87] | Biological production in mammalian cell cultures [100] |
| Scalability | High; relatively easy to scale up, as demonstrated during the COVID-19 pandemic [3] [103] | Moderate to Low; complex and costly to scale; faces technical bottlenecks in yield and purification [100] [102] |
| Primary Cost Drivers | Specialized lipid raw materials, formulation optimization, analytical characterization [103] [99] | Advanced GMP facility overhead, costly cell culture media, extensive purification and quality control steps [100] [102] |
| Manufacturing Timeline | Shorter production cycles; suitable for rapid response [3] | Lengthy production cycles; often takes several weeks [104] |
| Key Manufacturing Hurdles | Achieving targeted organ tropism, ensuring long-term stability, regulatory path for novel lipids [103] [99] | Overcoming low vector yields, preventing contamination, managing immunogenicity, avoiding insertional mutagenesis [3] [100] [102] |
| Dependence on CDMOs | High; over 50 companies offer LNP development services [103] | High; CDMOs are critical for meeting global demand, especially for smaller biotechs [100] [102] |
Experimental Protocols for Manufacturing Assessment
Evaluating the scalability and cost of vector production requires robust experimental methodologies. Below are generalized protocols for key analytical processes.
Protocol 1: Assessing LNP Critical Quality Attributes (CQAs)
Objective: To characterize the physical and chemical properties of a formulated LNP batch, which are critical for process consistency and scalability [87].
Methodology:
- Particle Size and Polydispersity Index (PDI): Determine using Dynamic Light Scattering (DLS). Low PDI indicates a uniform particle population, which is a hallmark of a robust and scalable manufacturing process.
- Encapsulation Efficiency: Quantify using a Ribogreen fluorescence-based assay. The nucleic acid payload is mixed with the reagent before and after disruption of the LNPs with a detergent. The difference in signal calculates the percentage of encapsulated material, directly impacting potency and cost-effectiveness.
- Zeta Potential: Measure via Laser Doppler Velocimetry. This indicates the surface charge of the particles, which influences colloidal stability and in vivo behavior.
- Morphology: Visualize using Cryo-Electron Microscopy (Cryo-EM) to confirm a spherical, core-shell structure and the absence of aggregates.
Protocol 2: Quantifying Viral Vector Titer and Potency
Objective: To determine the concentration and functional activity of a viral vector batch, essential for dose consistency and process yield calculations [100] [102].
Methodology:
- Physical Titer (Vector Genomes/mL): Extract the vector genome from a purified sample and quantify using digital droplet PCR (ddPCR). This provides an absolute count of viral particles, critical for scaling production batches.
- Infectious Titer (Transducing Units/mL): Incubate serial dilutions of the vector with a permissive cell line (e.g., HEK293). After a suitable period, quantify the number of transduced cells (e.g., by flow cytometry for a fluorescent reporter protein). This measures the functional potency of the batch.
- Ratio of Physical to Infectious Titer: Calculate the ratio of vector genomes to transducing units. A ratio close to 1 indicates a highly potent and pure preparation, whereas a high ratio suggests a significant proportion of defective particles, highlighting inefficiencies in the production or purification process.
Logical Workflow and Signaling Pathways in Vector Production
The following diagram illustrates the key decision points and comparative pathways in selecting and scaling a vector manufacturing platform.
Diagram Title: Decision Workflow for Vector Manufacturing Platform Selection.
The Scientist's Toolkit: Key Research Reagents and Materials
Successful development and scalable production of gene delivery vectors require a suite of specialized reagents and materials.
Table 3: Essential Research Reagents for Vector Development and Analysis
| Reagent/Material | Function | Application Context |
|---|---|---|
| Ionizable Lipids | Key functional lipid for nucleic acid encapsulation and endosomal escape [87]. | LNP Formulation |
| DMG-PEG 2000 | PEG-lipid that confers stability and controls particle size during LNP formation [103]. | LNP Formulation |
| Plasmid DNA (pDNA) | Genetic template for in vitro transcription of mRNA or for transfection in viral vector production [100]. | LNP & Viral Vector Manufacturing |
| Suspension Cell Lines (e.g., HEK293) | Scalable host cells for the production of viral vectors in bioreactors [102]. | Viral Vector Upstream Processing |
| Affinity Chromatography Resins | For purification of viral vectors based on specific tags or properties, crucial for yield and purity [102]. | Viral Vector Downstream Processing |
| Transfection Reagents | Chemicals or polymers to introduce genetic material (e.g., plasmid encoding the virus) into production cells [100]. | Viral Vector Upstream Processing |
| ddPCR/qPCR Reagents | For precise quantification of vector genome titer and monitoring of process residuals [102]. | Analytics & QC |
| Dynamic Light Scattering (DLS) Instrument | For measuring LNP particle size, distribution (PDI), and zeta potential [87]. | Analytics & QC |
The choice between lipid nanoparticles and viral vectors for gene delivery is not merely a biological question but a strategic one dictated heavily by manufacturing and economic realities. Lipid nanoparticles offer a distinct advantage in scalability, production speed, and cost-effectiveness for applications requiring transient expression, such as vaccines or short-term therapies [3] [99]. Their synthetic nature and lower immunogenicity also favor re-dosing strategies. Conversely, viral vectors remain indispensable for therapies demanding high-efficiency delivery and long-lasting or permanent gene expression, despite their complex and costly manufacturing processes [3] [100] [102].
The future landscape will likely be shaped by innovations aimed at overcoming the limitations of each platform. For LNPs, this involves developing novel lipids for improved tissue targeting and safety profiles [103] [87]. For viral vectors, advances in bioprocessing, automation, and the use of AI to optimize yields are critical to reduce costs and improve scalability [100] [102]. Ultimately, the decision must integrate therapeutic goals with a clear-eyed assessment of the production complexity and cost, ensuring that groundbreaking gene therapies can be manufactured reliably and accessibly for patients worldwide.
The choice between lipid nanoparticles (LNPs) and viral vectors is fundamental in gene therapy, dictiating the type of genetic cargo that can be delivered and the resulting therapeutic outcome. The table below provides a high-level comparison of their core characteristics.
| Feature | Lipid Nanoparticles (LNPs) | Viral Vectors (e.g., AAV, Lentivirus) |
|---|---|---|
| Primary Cargo Types | mRNA, siRNA, CRISPR RNP, plasmid DNA [3] [105] [106] | DNA, CRISPR-Cas9 components, shRNA [1] |
| Typical Payload Size | Versatile; no strict inherent limit [3] | Limited (e.g., AAV: ~4.7 kb [106]) |
| Expression Duration | Transient (days to weeks) [3] | Long-term to permanent [3] [1] |
| Key Advantages | Lower immunogenicity, suitable for repeated dosing, scalable production, rapid manufacturing [3] | High delivery efficiency, excellent tissue targeting, sustained gene expression [3] [1] |
| Major Limitations | Lower efficiency in some tissues, endosomal escape is a major barrier [28] | Risk of immunogenicity and insertional mutagenesis, complex and costly production [3] [1] |
In-Depth Cargo Analysis and Performance Data
mRNA and siRNA Delivery
LNPs are the established leader for delivering mRNA and siRNA, as demonstrated by their clinical success in COVID-19 vaccines and the siRNA therapeutic Onpattro [105] [51]. Their versatility allows for the delivery of both large (mRNA) and small (siRNA) RNA molecules.
Experimental Insight: Research reveals that LNPs trigger endosomal membrane damage, marked by galectin-9 recruitment, which is conducive to the cytosolic release of RNA [28]. However, this process is inefficient, with only a small fraction of the RNA cargo successfully escaping into the cytosol, highlighting a significant barrier to higher efficacy [28].
CRISPR-Cas9 Genome Editing Tools
Delivering the CRISPR-Cas9 system is a key test for any gene delivery vehicle, as it requires the simultaneous delivery of multiple components.
Comparative Experimental Data: A direct comparative study investigated LNP-mediated delivery of CRISPR-Cas9 in two forms: (1) as an mRNA (encoding Cas9) plus a single-guide RNA (sgRNA), and (2) as a preassembled ribonucleoprotein complex (RNP) [107].
- Editing Efficiency: In vitro and in vivo, LNPs delivering mRNA Cas9/sgRNA demonstrated higher gene editing efficiencies. In mice, this formulation achieved 60% gene knock-out in hepatocytes, whereas the RNP formulation did not lead to detectable in vivo editing under the tested conditions [107].
- Biodistribution: Systemic administration revealed that while mRNA Cas9 LNPs were retained mainly in the liver, Cas9 RNP LNPs showed a broader distribution, with additional accumulation in the spleen and lungs [107].
Plasmid DNA (pDNA) Delivery
While viral vectors are traditionally used for DNA delivery, recent advances are enabling LNP-based pDNA delivery, which offers the potential for prolonged transgene expression compared to mRNA.
Experimental Insight: A high-throughput screening of over 1,000 LNP formulations identified optimized pDNA-LNPs for liver-targeted expression. The study found that the choice of helper lipid (e.g., cationic DOTAP) and the ratios of LNP components were critical for efficient in vivo transfection. Furthermore, co-delivery of pDNA with siRNAs targeting inflammatory pathways (STAT, NF-κB) was shown to substantially extend the duration of transgene expression by reducing immune-mediated silencing [108].
Key Methodologies from Cited Research
- Objective: To compare the gene editing efficiency of LNPs encapsulating Cas9 mRNA/sgRNA versus Cas9 RNP.
- LNP Formulation: CRISPR components were encapsulated in LNPs using microfluidic mixing. Standard lipid components (ionizable lipid, phospholipid, cholesterol, PEG-lipid) were used.
- In Vitro Testing: Formulations were tested on reporter HEK293T and HEPA 1–6 cells. Editing efficiency was quantified using flow cytometry or other relevant assays.
- In Vivo Testing: Formulations were administered systemically to Ai9 reporter mice.
- Biodistribution Analysis: Tissues (liver, spleen, lungs) were harvested and analyzed for LNP presence.
- Gene Editing Assessment: Gene knock-out efficiency in hepatocytes was quantified, for example, by sequencing the target locus.
- Objective: To identify optimal LNP formulations for liver-targeted pDNA delivery from a large library.
- Library Design: A library of 1080 LNP formulations was designed by varying: ionizable lipid-to-helper lipid ratio, cholesterol-to-PEG-lipid ratio, total lipid percentage, and N/P ratio. Six helper lipids with different charges were tested.
- High-Throughput In Vitro Screening: The library was screened for transfection efficiency (using luciferase pDNA) and cytotoxicity in HepG2 liver cells.
- Cluster-Mode In Vivo Screening: Top-performing formulations were grouped into clusters. Clusters were screened via intrahepatic injection in mice to assess local transfection and toxicity.
- Final Validation: The most promising clusters from the intrahepatic screen were advanced to systemic intravenous administration to identify the top individual formulations for liver-targeted gene expression.
Visualizing the Delivery Pathways and Workflows
Diagram 1: Fundamental Mechanisms of Gene Delivery
Diagram 2: Systematic Screening of LNP Formulations
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function / Role in Research |
|---|---|
| Ionizable Cationic Lipid (e.g., MC3) | The key functional component of LNPs; promotes nucleic acid complexation, cellular uptake, and endosomal escape [105] [109]. |
| Helper Lipids (DSPC, DOPE) | Phospholipids that enhance LNP stability and membrane integrity; can influence fusogenicity and endosomal escape [109]. |
| PEGylated Lipid | Improves nanoparticle stability and circulation time by reducing aggregation and non-specific protein adsorption [109]. |
| Adeno-Associated Virus (AAV) | A viral vector prized for in vivo gene therapy due to its low immunogenicity and long-term gene expression profile [1] [106]. |
| Lentivirus | An integrating viral vector ideal for ex vivo gene therapy applications requiring stable, long-term transgene expression in dividing cells [1] [106]. |
| Reporter Genes (Luciferase, GFP) | Encoded by delivered DNA or mRNA to quantitatively (luciferase) or qualitatively (GFP) assess transfection/transduction efficiency [107] [108]. |
| Electroporation Systems | A physical non-viral method using electrical pulses to create pores in cell membranes for nucleic acid delivery, commonly used ex vivo [106]. |
The success of any gene therapy product is fundamentally dependent on the selection of an appropriate delivery vector. This choice dictates critical factors including therapeutic efficacy, safety, manufacturing feasibility, and commercial viability. The two leading technologies—viral vectors and lipid nanoparticles (LNPs)—each possess distinct strengths and limitations. This guide provides an objective, data-driven comparison to inform researchers and drug development professionals in selecting the optimal platform based on specific therapeutic goals, target tissues, and genetic cargo requirements.
Platform Comparison at a Glance
The table below summarizes the core technical and clinical characteristics of the dominant viral vectors and LNPs to provide a high-level strategic overview.
Table 1: Head-to-Head Comparison of Gene Delivery Platforms
| Feature | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Lipid Nanoparticles (LNP) |
|---|---|---|---|
| Primary Use Case | In vivo gene replacement (CNS, Eye, Liver) [110] | Ex vivo cell therapy (CAR-T, HSCs) [110] | Gene editing (CRISPR/mRNA), Vaccines [110] |
| Cargo Capacity | ~4.7 kb (Strict) [110] | ~10 kb (Moderate) [110] | Flexible / High (Virtually unlimited for nucleic acids) [110] [2] |
| Genetic Persistence | Episomal (Long-term in non-dividing cells) [110] | Integrated (Permanent in dividing cells) [110] | Transient (Ideal for editing) [110] |
| Immunogenicity | High (Pre-existing NAbs prevent re-dosing) [110] | Low (Use is mostly ex vivo) [110] | Low (Suitable for re-dosing) [3] [110] |
| Key Safety Concerns | Immune reactions, potential hepatotoxicity [3] | Insertional mutagenesis (theoretical risk) [110] | Acute inflammation (for pDNA), carrier-related toxicities [111] |
| Manufacturing COGS | High (Complex cell culture & purification) [110] | High (Shear sensitivity, low yield) [110] | Low to Medium (Scalable chemical synthesis) [110] |
| Key CMC Bottleneck | Empty/Full Capsid Separation [110] | Viral Stability & Titer [110] | Lipid Purity & Microfluidic Fouling [110] |
Detailed Analysis by Therapeutic Intent
For Long-Term Gene Replacement or Addition
Recommended Platform: Adeno-Associated Virus (AAV)
- Mechanism of Action: AAV vectors are engineered to be replication-deficient and deliver their genetic payload into the nucleus of target cells, where it persists as a stable episome, leading to durable protein expression [3] [110].
- Supporting Data: AAV is the established standard for in vivo treatments of monogenic disorders affecting post-mitotic tissues. Therapies like Luxturna for a genetic retinal disease demonstrate its clinical success [9].
- Critical Considerations:
- Cargo Limit: The strict ~4.7 kb capacity is a major constraint for delivering large genes [110].
- Immunogenicity: Pre-existing neutralizing antibodies (NAbs) can exclude a significant portion (30-60%) of the patient population from treatment and prevent effective re-dosing [110].
- Targeting: Specific AAV serotypes (e.g., AAV9, AAVrh74) and engineered capsids offer known tissue tropism, enabling delivery to the retina, central nervous system, and liver [110].
For PermanentEx VivoCell Engineering
Recommended Platform: Lentivirus (LV)
- Mechanism of Action: Lentiviral vectors can integrate their genetic payload into the host genome of dividing cells. This ensures the therapeutic gene is passed on to daughter cells, enabling permanent genetic modification [3] [110].
- Supporting Data: LV is the backbone of the cell therapy revolution, including CAR-T therapies for blood cancers. Its larger ~10 kb cargo capacity allows for complex payloads, such as a Chimeric Antigen Receptor (CAR) plus safety switches [110] [9].
- Critical Considerations:
- Safety: While self-inactivating (SIN) vectors have reduced the risk, the theoretical possibility of insertional mutagenesis necessitates long-term patient follow-up studies [110].
- Application: Used almost exclusively in ex vivo settings where cells are modified outside the body before reinfusion.
For Transient Expression: Vaccines, Gene Editing, and Protein Replacement
Recommended Platform: Lipid Nanoparticles (LNP)
- Mechanism of Action: LNPs fuse with the cell membrane, delivering their payload (e.g., mRNA, siRNA) directly into the cytoplasm. This results in rapid but transient protein expression, as the RNA is not integrated into the genome and degrades naturally [3] [2].
- Supporting Data: The success of COVID-19 mRNA vaccines validated LNP performance. LNPs are the preferred vehicle for CRISPR-Cas9 components, where transient activity is desired to minimize off-target effects [110] [35] [87].
- Critical Considerations:
- Biodistribution: Systemically administered LNPs naturally accumulate in the liver. Targeting extra-hepatic tissues requires complex lipid chemistry that is still in development [110].
- Inflammation: Plasmid DNA (pDNA)-LNPs can trigger acute, dose-limiting inflammation via the cGAS-STING pathway. Strategies to mitigate this, such as loading the STING inhibitor nitro-oleic acid (NOA), are showing promise in preclinical studies [111].
- Re-dosability: Low immunogenicity compared to viral vectors allows for multiple administrations, a key advantage for treatments requiring repeated dosing [3] [110].
Essential Experimental Protocols and Workflows
Protocol: Enhancing Viral Vector Transfection with Transportan Peptide
This simple co-administration protocol can improve gene delivery efficiency, particularly for difficult-to-transfect cell lines and primary cells [44].
- Preparation: Synthesize or obtain high-titer GFP-expressing viral vectors (Lentivirus or AAV) and Transportan (TP) peptide (sequence: GWTLNSAGYLLGKINLKALAALAKKIL).
- Cell Seeding: Plate the target cells (e.g., PC3, Raw264.7, BMDMs, RPE) at an appropriate density and allow them to adhere.
- Transfection Mixture: Dilute the GFP viral vector in a serum-free medium. Add TP peptide directly to this dilution to achieve a final working concentration (e.g., 5-10 µM). Mix gently by pipetting.
- Co-incubation: Remove growth medium from cells and add the virus-TP mixture. Incubate cells with this mixture for 48 hours.
- Analysis: Assess transfection efficiency via fluorescent imaging for GFP expression and/or flow cytometry analysis [44].
Diagram 1: TP enhancement workflow
Protocol: Mitigating pDNA-LNP-Induced Inflammation via STING Inhibition
This methodology outlines the formulation of safer pDNA-LNPs by incorporating the anti-inflammatory lipid NOA [111].
- LNP Formulation: Prepare pDNA-LNPs using standard microfluidic mixing techniques. Common ionizable lipids include ALC-0315, D-Lin-MC3-DMA, or SM-102.
- NOA Loading: Co-dissolve nitro-oleic acid (NOA) with the other lipid components (ionizable lipid, cholesterol, helper lipid, PEG-lipid) in an organic phase prior to mixing with the aqueous pDNA solution.
- Buffer Exchange & Characterization: After mixing, dialyze or use tangential flow filtration to remove residual solvent and place LNPs in the final buffer. Characterize particles for size, PDI, and encapsulation efficiency.
- In Vitro Validation: Treat macrophage cell lines (e.g., RAW264.7) with NOA-pDNA-LNPs and standard pDNA-LNPs. Measure cell viability and secretion of pro-inflammatory cytokines (IFN-β, IL-6) to confirm reduced immunogenicity.
- In Vivo Validation: Administer NOA-pDNA-LNPs intravenously to naive C57BL/6 mice and monitor for acute inflammatory responses, mortality, and compared plasma cytokine levels against controls [111].
Diagram 2: pDNA-LNP STING pathway mechanism
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Gene Delivery Research
| Reagent / Solution | Function / Application | Key Considerations |
|---|---|---|
| AAV Serotypes (e.g., AAV2, AAV9, AAVrh74) | Delivery to specific tissues (CNS, liver, muscle). Well-trodden regulatory path [110]. | Pre-screen patients for pre-existing neutralizing antibodies. |
| Lentiviral Vectors (3rd Generation, SIN) | Stable genetic modification of dividing cells ex vivo (e.g., HSCs, T-cells) [110]. | Requires long-term follow-up due to theoretical risk of insertional mutagenesis. |
| Ionizable Lipids (e.g., ALC-0315, SM-102) | Core component of LNPs; enables encapsulation and endosomal escape [87] [111]. | Lipid structure and pKa critically determine in vivo performance and tropism [88]. |
| Transportan (TP) Peptide | Cell-penetrating peptide to enhance viral vector uptake via macropinocytosis [44]. | Simple co-incubation protocol; effective in primary and difficult-to-transfect cells. |
| Nitro-Oleic Acid (NOA) | Endogenous anti-inflammatory lipid; inhibits STING pathway in pDNA-LNPs [111]. | Mitigates acute inflammation, enabling safer in vivo pDNA delivery. |
| PEGylated Lipids | Stabilizes LNPs, reduces immune clearance, and increases circulation half-life [2]. | Can induce Accelerated Blood Clearance (ABC) phenomenon upon repeated dosing. |
Emerging Trends and Future Outlook
The field is moving towards hybrid and platform approaches. Regulatory agencies are increasingly open to treating proven delivery vehicles as "platforms," potentially reducing the preclinical burden for new payloads [110]. Computationally driven design, using molecular dynamics and machine learning, is accelerating the optimization of LNP formulations and the engineering of novel viral capsids [88]. Furthermore, non-viral delivery of plasmid DNA via improved LNPs is emerging as a promising strategy for achieving longer-term transgene expression while maintaining the safety and manufacturing advantages of non-viral systems [111]. The most successful R&D pipelines are becoming "delivery-agnostic," selecting the optimal vector based on a precise match between therapeutic intent and platform capabilities.
Conclusion
The choice between lipid nanoparticles and viral vectors is not a quest for a universal winner, but a strategic decision based on therapeutic requirements. Viral vectors remain unparalleled for long-term, high-efficiency gene expression in specific tissues, while LNPs excel in safety, scalability, and versatile delivery of various nucleic acids for transient expression. Future directions point towards hybrid solutions and increasingly sophisticated engineering—such as cell-specific LNPs and refined viral capsids—to overcome biological barriers. This progression will expand the reach of gene therapies from rare diseases to broader clinical indications, solidifying gene delivery as a cornerstone of modern medicine.
References
- 1. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11242246/]
- 2. Lipid Nanoparticles for Gene Delivery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5006671/]
- 3. Lipid Nanoparticles vs. Viral Vectors: A Comparison for ... [https://www.helixbiotech.com/post/lipid-nanoparticles-vs-viral-vectors-a-comparison-for-gene-therapy]
- 4. The next frontier of in vivo gene therapy: are LNPs the new ... [https://www.darkhorseconsultinggroup.com/post/the-next-frontier-of-in-vivo-gene-therapies]
- 5. Are LNPs the new AAV? [https://www.cellgenetherapyreview.com/3975-Featured-Articles/621087-Are-LNPs-the-new-AAV/]
- 6. Hurdles to healing: Overcoming cellular barriers for viral ... [https://www.sciencedirect.com/science/article/pii/S0378517325003060]
- 7. Advancing Gene Delivery LNPs Adenovirus Lentivirus And ... [https://www.cellandgene.com/doc/advancing-gene-delivery-lnps-adenovirus-lentivirus-and-more-0002]
- 8. Emerging non-viral vectors for gene delivery [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-023-02044-5]
- 9. Viral and non-viral vectors in gene therapy: current state ... [https://www.sciencedirect.com/science/article/pii/S2352396425002786]
- 10. Directed evolution of engineered virus-like particles with ... [https://www.nature.com/articles/s41587-024-02467-x]
- 11. Is the tide turning on viral vectors? The shifting landscape ... [https://www.bioprocessintl.com/therapeutic-class/is-the-tide-turning-on-viral-vectors-the-shifting-landscape-of-gene-therapy-delivery]
- 12. Viral and non-viral vectors in gene therapy: current state and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12271757/]
- 13. Non-viral vectors in gene therapy: LNPs in the spotlight [https://www.susupport.com/blogs/knowledge/non-viral-vectors-gene-therapy-lnps-spotlight]
- 14. DNA-LNPs: A safer, longer-lasting gene therapy ... [https://www.pennmedicine.org/news/dna-lnps-a-safer-longer-lasting-gene-therapy-breakthrough]
- 15. Viral Vector Manufacturing Market to Surpass USD 10.65 [https://www.globenewswire.com/news-release/2025/11/24/3193529/0/en/Viral-Vector-Manufacturing-Market-to-Surpass-USD-10-65-Billion-by-2033-Driven-by-Surging-Gene-Therapy-Pipelines-and-Advanced-Vector-Engineering-SNS-Insider.html]
- 16. Artificial Intelligence-Driven Strategies for Targeted Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389219/]
- 17. A versatile antibody capture system drives specific in vivo ... [https://www.nature.com/articles/s41565-025-01954-9]
- 18. Long-term stability and immunogenicity of lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11085994/]
- 19. Advancing gene editing: the role of lipid nanoparticles in ... [https://www.drugtargetreview.com/article/188056/advancing-gene-editing-the-role-of-lipid-nanoparticles-in-crispr-delivery/]
- 20. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 21. The State and Future of Viral, Nonviral Vectors for Gene ... [https://www.ajmc.com/view/the-state-and-future-of-viral-non-viral-vectors-for-gene-therapy]
- 22. Engineering of mRNA vaccine platform with reduced lipids ... [https://www.nature.com/articles/s41467-025-63965-3]
- 23. Transduction vs Transfection: Understanding Gene ... [https://www.assaygenie.com/blog/transduction-vs-transfection?srsltid=AfmBOooCdPc0ruJfCaVVlARx_O33CKIEo4aeeTGUN5QzWkdeJeSx1xIy]
- 24. Gene Delivery Decoding: Viral Transduction vs. ... [https://www.cd-genomics.com/biomedical-ngs/resource/gene-delivery-viral-transduction-transfection-applicability.html]
- 25. Transfection Technologies for Next-Generation Therapies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347970/]
- 26. What Is Transformation, Transfection & Transduction? [https://www.zymoresearch.com/blogs/blog/what-is-transformation-transfection-transduction?srsltid=AfmBOorrPuAFh6MQVSCkjyXUwJZ5SfM7VozF__s31Oihk0Cg4hu53roc]
- 27. What is the difference between transfection and ... [https://www.mirusbio.com/difference-transfection-transduction/?srsltid=AfmBOoqrTWKPgdVCatKg7Sp2wR_7ilPr3j-ZpfWylbx6h4DbY_c-5QuJ]
- 28. Cellular and biophysical barriers to lipid nanoparticle ... [https://www.nature.com/articles/s41467-025-60959-z]
- 29. Lipid Nanoparticle Applications in Gene Editing Therapies [https://www.precigenome.com/post/lipid-nanoparticle-applications-in-gene-editing-therapies?srsltid=AfmBOorbGEMog9JLEbMdXf9q0U2Rmd_op46qf_flf0yVeG25Rtkh1caJ]
- 30. Advancing gene editing therapeutics: Clinical trials and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12395446/]
- 31. Membrane-modified lipid nanoparticles for RNA delivery [https://www.sciencedirect.com/science/article/pii/S2329050125001007]
- 32. High‐Throughput Strategies for Streamlining Lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622538/]
- 33. Engineering viral vectors for acoustically targeted gene ... [https://www.nature.com/articles/s41467-024-48974-y]
- 34. AAV vector development, back to the future [https://www.sciencedirect.com/science/article/pii/S1525001625002734]
- 35. The Hope for Lipid-Based Nanoparticles in Blood Disorders [https://www.sciencedirect.com/science/article/pii/S2452318625000546]
- 36. Designing lipid nanoparticles using a transformer-based ... [https://www.nature.com/articles/s41565-025-01975-4]
- 37. Case studies [https://bgtcplaybook.document360.io/docs/case-studies-1]
- 38. Clinical and pharmacovigilance safety evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616891/]
- 39. innovations and challenges in rAAV vector-based CRISPR ... [https://www.nature.com/articles/s41434-025-00573-2]
- 40. Progress and challenges in viral vector manufacturing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4802372/]
- 41. Lipid Nanoparticle Applications in Gene Editing Therapies [https://www.precigenome.com/post/lipid-nanoparticle-applications-in-gene-editing-therapies?srsltid=AfmBOoq27iU6WC2gLU6cq0FWt9DgopkzWI6W-LfkR2CP6afnQ-tQs75E]
- 42. T cell-specific non-viral DNA delivery and in vivo CAR-T ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12258353/]
- 43. Emerging Approaches for the Discovery of Lipid-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473667/]
- 44. Improving viral-based gene delivery to mammalian cells ... [https://www.sciencedirect.com/science/article/abs/pii/S0022354925003727]
- 45. Intellia Announces Positive Clinical Proof-of-Concept Data ... [https://ir.intelliatx.com/news-releases/news-release-details/intellia-announces-positive-clinical-proof-concept-data-redosing]
- 46. Acuitas Therapeutics Unveils Next-Generation Lipid ... [https://crisprmedicinenews.com/press-release-service/card/acuitas-therapeutics-unveils-next-generation-lipid-nanoparticle-advancements-at-the-2025-mrna-health/]
- 47. Nanoparticle-based strategy in CAR-T cell immunotherapy [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02476-7]
- 48. Viral and non-viral vectors in gene therapy: current state ... [https://pubmed.ncbi.nlm.nih.gov/40602323/]
- 49. T cell-specific non-viral DNA delivery and in vivo CAR ... [https://pubmed.ncbi.nlm.nih.gov/40659448/]
- 50. Lipid Nanoparticles: A Novel Gene Delivery Technique for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9600891/]
- 51. Lipid nanoparticles for mRNA delivery [https://www.nature.com/articles/s41578-021-00358-0]
- 52. Viral vector platforms within the gene therapy landscape [https://www.nature.com/articles/s41392-021-00487-6]
- 53. Viral Vectors in Gene Therapy - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6023384/]
- 54. Novel Insights into the Therapeutic Potential of Lung- ... [https://www.mdpi.com/2073-4409/11/6/984]
- 55. Past, present, and future of liver-directed gene therapy [https://www.sciencedirect.com/science/article/pii/S1525001625002151]
- 56. Current landscape of cystic fibrosis gene therapy [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1476331/full]
- 57. Enhancing RNA-lipid nanoparticle delivery: Organ [https://www.sciencedirect.com/science/article/pii/S0168365924005765]
- 58. Beyond the Virus: How Non-Viral Vectors Are Changing Gene ... [https://us.progen.com/post/Beyond-the-Virus-How-Non-Viral-Vectors-Are-Changing-Gene-Therapy]
- 59. Intranasal and Pulmonary Lipid Nanoparticles for Gene Delivery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204099/]
- 60. Advances in Inhaled Nanoparticle Drug Delivery for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12598920/]
- 61. Charge-assisted stabilization of lipid nanoparticles enables ... [https://www.nature.com/articles/s41467-024-53914-x]
- 62. Optimized inhaled LNP formulation for enhanced treatment ... [https://www.nature.com/articles/s41467-024-51056-8]
- 63. Immunogenicity of lipid nanoparticles and its impact on the ... [https://www.nature.com/articles/s12276-023-01086-x]
- 64. The mRNA component of LNP-mRNA vaccines triggers ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1670350/full]
- 65. Integration of TLR7/8 agonists into lipid nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629014/]
- 66. Lung and liver editing by lipid nanoparticle delivery of a ... [https://www.nature.com/articles/s41587-024-02437-3]
- 67. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://www.mdpi.com/1422-0067/25/13/7333]
- 68. Size control of lipid nanoparticles via simulation-based design ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03836-7]
- 69. Advancing AAV Vector Manufacturing: Challenges, ... [https://www.frontiersin.org/journals/molecular-medicine/articles/10.3389/fmmed.2025.1709095/full]
- 70. Lipid nanoparticles: a promising tool for nucleic acid delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12328551/]
- 71. Driving forces in the assembly of lipid nanoparticles ... [https://www.nature.com/articles/s41598-025-20340-y]
- 72. Past, Present, and Future of Viral Vector Vaccine Platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12115715/]
- 73. Non-Viral Gene Delivery Systems: Current Advances and ... [https://pubmed.ncbi.nlm.nih.gov/40874945/]
- 74. Gene Therapy Breakthrough: New Evidence Shows ... [https://globalrph.com/2025/04/gene-therapy-breakthrough-new-evidence-shows-promise-for-hemophilia-cure/]
- 75. Lipid nanoparticles for mRNA delivery in brain via systemic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346281/]
- 76. Why do lipid nanoparticles target the liver? Understanding ... [https://www.sciencedirect.com/science/article/pii/S2329050125000312]
- 77. Lipid Nanoparticles LNP Market Outlook 2025-2032 [https://www.intelmarketresearch.com/lipid-nanoparticles-2025-2032-904-965]
- 78. Formulation-Driven Optimization of PEG-Lipid Content in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388858/]
- 79. Optimizing the targeting of lipid nanoparticles for gene therapy [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00351b]
- 80. Impact of ionizable lipid type on the pharmacokinetics and ... [https://pubmed.ncbi.nlm.nih.gov/40505892/]
- 81. Impact of ionizable lipid type on the pharmacokinetics and ... [https://www.sciencedirect.com/science/article/pii/S0168365925005656]
- 82. Lipid Nanoparticle News and Research Summary (August 19 [https://www.precigenome.com/post/lipid-nanoparticle-news-and-research-summary-august-19-september-10-2025?srsltid=AfmBOopPKsoxpqA9tnLjAzWj6DSEUED_BP0c5_8uh6LmtHlk2x2iCUHV]
- 83. PEGylated lipids in lipid nanoparticle delivery dynamics ... [https://pubmed.ncbi.nlm.nih.gov/41189930/]
- 84. Engineering Lipid Nanoparticles for mRNA Immunotherapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC11976204/]
- 85. Choosing the Right Vector for Gene Therapy Applications [https://blog.mblintl.com/choosing-the-right-vector-for-gene-therapy-applications]
- 86. Particles that enhance mRNA delivery could reduce ... [https://news.mit.edu/2025/particles-enhance-mrna-delivery-could-reduce-vaccine-dosage-costs-1107]
- 87. Review Lipid nanoparticles: Composition, formulation, and ... [https://www.sciencedirect.com/science/article/pii/S2329050125000580]
- 88. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 89. Appraisal for the Potential of Viral and Nonviral Vectors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9407213/]
- 90. Delivering the Message: Translating mRNA Therapy for Liver ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12361847/]
- 91. Lipid nanoparticle (LNP) mediated mRNA delivery in ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365924003717]
- 92. Lipid Nanoparticle (LNP) Drug Delivery Advances March 4- ... [https://www.precigenome.com/post/lnplipid-nanoparticle-drug-delivery-advances-march-4-10-2025-1?srsltid=AfmBOoouG9xppgzojRjDtmVuMWj07Cq-m4acd6Q484gajs1hWlFkXfXD]
- 93. Novel Viral Vector Manufacturing Approach Could Improve ... [https://pediatricsnationwide.org/2025/05/15/novel-viral-vector-manufacturing-approach-could-improve-safety-of-gene-therapy/]
- 94. Improved localized mRNA delivery using lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606572/]
- 95. Optimizing viral transduction in immune cell therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046913/]
- 96. LIPID NANOPARTICLE COMPOSITION AND ... [https://www.sciencedirect.com/science/article/pii/S1465324925001446]
- 97. Quantitative Comparison of Intracellular Unpacking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3014860/]
- 98. Retroviral Vector Technology for Gene Therapy [https://www.sciencedirect.com/science/article/pii/S002228362500539X]
- 99. Lipid Nanoparticles Market Size, Share Analysis, 2025-2032 [https://www.coherentmi.com/industry-reports/lipid-nanoparticles-market]
- 100. Viral Vector Manufacturing Research Report 2025 with ... [https://finance.yahoo.com/news/viral-vector-manufacturing-research-report-144500354.html]
- 101. United States Lipid Nanoparticle Market Forecast and ... [https://finance.yahoo.com/news/united-states-lipid-nanoparticle-market-105100859.html]
- 102. Viral Vector Manufacturing Market Trends and Future ... [https://www.congruencemarketinsights.com/report/viral-vector-manufacturing-market]
- 103. LNP manufacturing market set to take off in the next decade [https://www.bioxconomy.com/modalities/lnp-manufacturing-market-set-to-take-off-in-the-next-decade]
- 104. Lipid nanoparticles for engineering next generation CAR T ... [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00432b]
- 105. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9176214/]
- 106. Exploring Delivery Methods for Gene Therapy Manufacturing [https://www.thermofisher.com/blog/behindthebench/unlocking-the-future-exploring-viral-and-non-viral-delivery-methods-for-gene-therapy-manufacturing/]
- 107. Comparative analysis of lipid Nanoparticle-Mediated ... [https://www.sciencedirect.com/science/article/pii/S093964112400033X]
- 108. Multi-step screening of DNA/lipid nanoparticles and co- ... [https://www.nature.com/articles/s41467-022-31993-y]
- 109. Recent Advances in the Lipid Nanoparticle-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10059764/]
- 110. Viral vs. Non-Viral Gene Delivery: A Critical Choice for ... [https://www.drugdiscoverynews.com/viral-vs-non-viral-gene-delivery-a-critical-choice-for-gene-therapy-developers-16819]
- 111. Safer non-viral DNA delivery using lipid nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12235526/]