The CRISPR Search Engine: How DNA Data Storage and CRISPR-Cas Are Revolutionizing Biomedical Research
This article provides a comprehensive overview of CRISPR-powered search engines for DNA-based data storage, a cutting-edge fusion of synthetic biology and information technology.
The CRISPR Search Engine: How DNA Data Storage and CRISPR-Cas Are Revolutionizing Biomedical Research
Abstract
This article provides a comprehensive overview of CRISPR-powered search engines for DNA-based data storage, a cutting-edge fusion of synthetic biology and information technology. Aimed at researchers and drug development professionals, we explore the foundational principles, detailing how digital data is encoded into synthetic DNA and retrieved using CRISPR-Cas systems like Cas9 for sequence-specific targeting. We dissect the methodological workflows, from library preparation to sequence-guided data retrieval, and address critical troubleshooting aspects such as off-target effects and read/write fidelity. The analysis validates the technology by comparing it to traditional electronic storage and other molecular search methods, highlighting its unparalleled density, longevity, and potential for rapid in-molecular analytics. The conclusion synthesizes the transformative implications for creating searchable molecular archives of genomic, clinical, and research data.
Decoding the Future: The Convergence of CRISPR Technology and DNA Data Storage
The exponential growth of digital data is rapidly outpacing the capacity and longevity of conventional silicon-based storage. Current projections indicate global datasphere volume will exceed 175 zettabytes by 2025, while the fundamental physical limits of silicon bit density (approaching ~1 Tb/in²) and its degradation over time (10-20 year archival lifespan) present an existential crisis. This necessitates the exploration of molecular data storage, with DNA emerging as a leading candidate due to its ultra-high theoretical density (~215 PB/g) and millennial-scale stability. This application note details experimental protocols and reagent solutions central to a CRISPR-powered search engine framework for retrieving information encoded within synthetic DNA archives.
Quantitative Analysis of Storage Media Limits
Table 1: Comparative Analysis of Data Storage Technologies
| Parameter | Hard Disk Drive (HDD) | Solid-State Drive (SSD) | Magnetic Tape (LTO-9) | DNA Data Storage (Theoretical) |
|---|---|---|---|---|
| Areal Density | ~1 Tb/in² (current) | N/A (3D NAND layers) | ~0.5 Gb/in² | ~1 Exabyte/mm³ |
| Practical Archival Lifespan | 5-10 years | 10-20 years (data retention) | 15-30 years | 100s - 1000s of years |
| Energy Use (Access, W/TB/hr) | ~3-5 W/TB (idle) | ~0.5-1 W/TB (idle) | ~0 W/TB (offline) | ~0 W/TB (offline storage) |
| Current Cost ($/TB) | $15-20 | $40-60 | $5-10 (media) | ~$1,000,000+ (synthesis) |
| Read Speed (Sequencing) | ~200 MB/s | ~500 MB/s | ~400 MB/s | ~100-400 MB/s (PromethION) |
| Write Speed | ~200 MB/s | ~500 MB/s | ~300 MB/s | ~1-10 KB/s (oligo synthesis) |
Table 2: Key Limitations of Silicon-Based Storage Scaling
| Limiting Factor | Physical Constraint | Current State (2024) | Projected "Wall" |
|---|---|---|---|
| Superparamagnetic Limit | Thermal stability of magnetic grains | ~1-1.5 Tb/in² (HAMR/Microwave Assisted) | ~4-5 Tb/in² (est.) |
| NAND Flash Cell Size | Quantum tunneling leakage, cell-to-cell interference | 140-150+ layers (3D NAND) | ~500 layers / ~10 nm pitch (est.) |
| Heat Dissipation | Energy per bit operation vs. chip thermal budget | ~20-30 W/cm² (advanced packages) | Fundamental cooling limit |
| Photolithography Wavelength | Extreme UV (EUV) lithography resolution | 13.5 nm wavelength, ~20 nm features | ~10-13 nm feature size limit |
Core Experimental Protocols for CRISPR-Powered DNA Data Retrieval
Protocol 2.1: Encoding and Synthesis of Data into DNA Oligo Pools
Objective: Convert digital binary data into nucleotide sequences, synthesize, and prepare for storage.
- Data Encoding: Use a robust encoding scheme (e.g., Fountain code-based DNA Fountain or RaptorQ codes) to convert binary files into nucleotide sequences (A, C, G, T). Implement error correction bits and sequence constraints (e.g., homopolymer limit ≤ 3, GC content 40-60%).
- Oligo Design: Append universal PCR priming sites (e.g., P5/P7) and a unique addressing crRNA-targetable barcode (20-30 nt) to each data-containing sequence. Final oligo length: 150-200 nt.
- Synthesis & Pooling: Utilize high-throughput phosphoramidite synthesis (e.g., on an Agilent SurePrint or Twist Bioscience platform). Synthesize all oligos in a single pooled library.
- Quality Control: Quantify pool via qPCR. Validate a random sample by next-generation sequencing (NGS, MiSeq) to confirm sequence fidelity and representation.
Protocol 2.2: Construction of dCas9-Based Searchable DNA Library
Objective: Clone the oligo pool into a plasmid vector to create a searchable "DNA data archive" using a nuclease-deficient CRISPR system.
- Vector Preparation: Linearize a high-copy plasmid (e.g., pUC19) containing a dead Cas9 (dCas9) gene under an inducible promoter (e.g., pLtetO-1) and a multiple cloning site (MCS) downstream of dCas9.
- Golden Gate Assembly: Perform a Golden Gate assembly using BsaI sites to clone the entire oligo pool into the MCS of the linearized vector. Each data oligo becomes a "data record" flanked by priming sites and its unique barcode.
- Transformation & Library Amplification: Transform the assembly reaction into electrocompetent E. coli (e.g., NEB 10-beta). Plate on selective agar to ensure >10x coverage of library diversity. Harvest all colonies for maxiprep plasmid DNA library.
Protocol 2.3: Targeted Retrieval via Programmable dCas9-Guided Enrichment
Objective: Use a specific crRNA to guide dCas9 to a target barcode, physically enrich the corresponding plasmid, and PCR-amplify the retrieved data for sequencing and decoding.
- CRISPR Ribonucleoprotein (RNP) Complex Formation:
- Design and synthesize a crRNA complementary to the target data barcode.
- Incubate 200 nM purified dCas9 protein with 240 nM crRNA (1:1.2 molar ratio) in NEBuffer 3.1 at 25°C for 10 minutes.
- Binding and Enrichment Reaction:
- In a 50 µL reaction, combine:
- 100 ng of the plasmid DNA library.
- 20 µL of pre-formed dCas9:crRNA RNP complex.
- 1x Binding Buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl₂, 0.1 mM EDTA, 5% Glycerol).
- Incubate at 37°C for 60 minutes.
- In a 50 µL reaction, combine:
- Magnetic Pull-Down:
- Pre-bind a biotinylated anti-His antibody (dCas9 is His-tagged) to streptavidin magnetic beads.
- Add the beads to the binding reaction and incubate for 15 minutes at RT.
- Place tube on a magnet, discard supernatant, and wash beads 3x with Wash Buffer (Binding Buffer + 0.01% Triton X-100).
- Elution and PCR Recovery:
- Elute bound DNA by adding 50 µL of Elution Buffer (10 mM Tris-HCl pH 8.0, 0.05% SDS) and incubating at 65°C for 10 minutes.
- Perform PCR on the eluate using primers specific to the universal priming sites flanking the data region.
- Purify PCR product and submit for Sanger or Illumina MiSeq sequencing.
- Data Decoding: Convert the retrieved nucleotide sequences back to binary data using the inverse of the encoding scheme from Protocol 2.1.
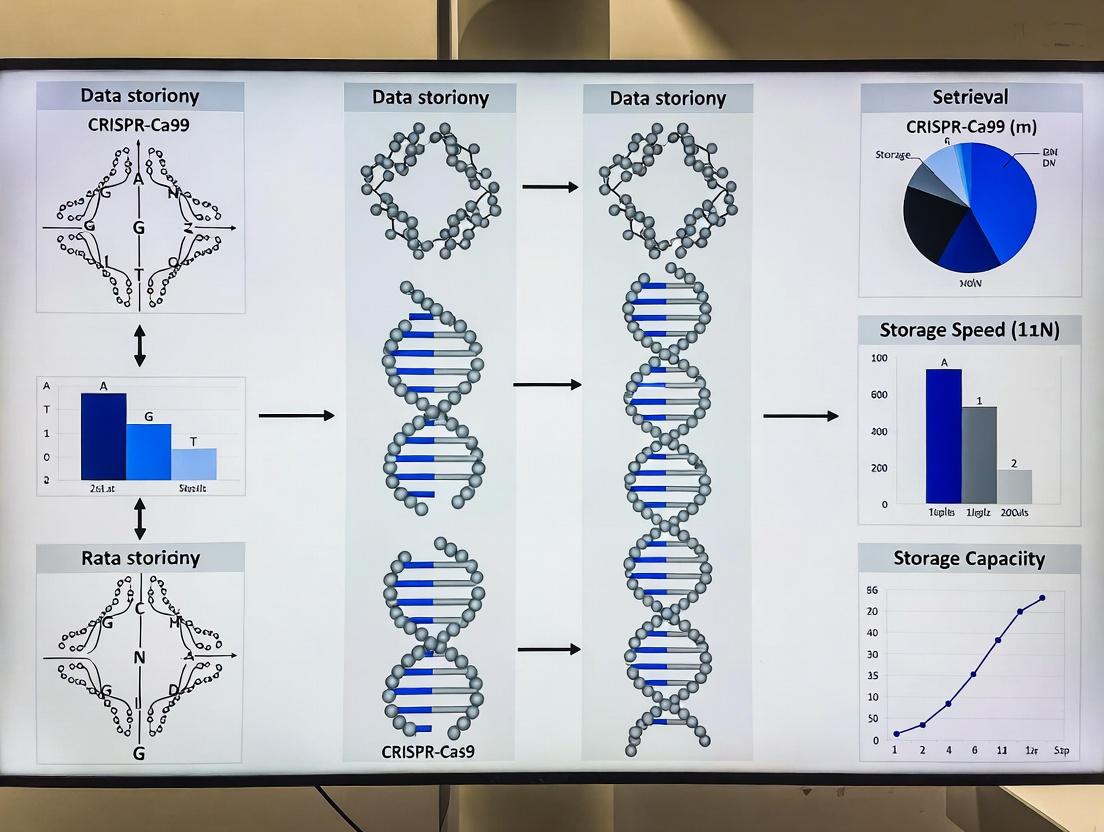
Diagrams for Experimental Workflow & System Logic
Title: DNA Data Storage and CRISPR Search Engine Workflow
Title: Molecular Mechanism of dCas9-Based DNA Data Retrieval
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-DNA Data Storage Experiments
| Reagent / Material | Provider Examples | Function in Protocol |
|---|---|---|
| dCas9 Protein (His-tagged) | IDT, Thermo Fisher, NEB | Core search engine protein; binds crRNA and target DNA without cleavage. His-tag enables magnetic enrichment. |
| Custom crRNA (ATTO 550 optional) | IDT, Synthego | Guides dCas9 to the specific barcode address of the target data. Fluorescent tag allows for validation via microscopy. |
| High-Competency E. coli | NEB (10-beta), Lucigen | Essential for efficient transformation and amplification of the large, complex plasmid data library. |
| Golden Gate Assembly Kit (BsaI) | NEB, Thermo Fisher | Modular, efficient cloning of the diverse oligo pool into the dCas9 plasmid vector. |
| Streptavidin Magnetic Beads | Thermo Fisher, MilliporeSigma | Solid-phase support for capturing the dCas9-DNA complex via biotin-avidin interaction. |
| Biotinylated Anti-His Antibody | Abcam, Thermo Fisher | Bridge between the His-tagged dCas9 and the streptavidin beads for magnetic pull-down. |
| Phusion High-Fidelity PCR Master Mix | Thermo Fisher, NEB | Amplifies the enriched target DNA with high fidelity prior to sequencing. |
| Next-Gen Sequencing Kit (MiSeq) | Illumina | Sequences the retrieved DNA oligos to convert biological data back to digital format. |
| Data-encoded DNA Oligo Pool | Twist Bioscience, Agilent | The synthetic DNA archive containing the encoded digital information. |
The exponential growth of global data necessitates revolutionary storage solutions. DNA data storage, encoding digital information into synthetic nucleotide sequences, presents a paradigm shift with unparalleled density (~215 PB/g) and longevity (centuries to millennia). The central challenge, however, lies in the efficient retrieval of specific data files from within a vast, complex molecular pool. This application note is framed within a broader thesis proposing a CRISPR-powered search engine for DNA data storage. This system conceptualizes the use of enzymatically inactive Cas9 (dCas9) proteins, guided by RNA sequences (gRNAs) corresponding to file addresses, to physically locate and flag target DNA strands for subsequent readout, enabling random-access and computationally assisted data retrieval.
Table 1: Comparison of Data Storage Media
| Medium | Areal Density (MB/mm²) | Volumetric Density (PB/g) | Lifespan (Years) | Read Speed (MB/s) | Write Speed (MB/s) |
|---|---|---|---|---|---|
| DNA (Theoretical) | 1.0E+8 | 215 | 1000+ | 0.1 - 10 | 0.001 - 0.01 |
| HDD (2023) | 1.5E-3 | ~0.0001 | 5-10 | 200 | 200 |
| SSD (NVMe) | 0.03 | ~0.001 | 5-10 | 7000 | 5000 |
| Magnetic Tape (LTO-9) | 0.02 | ~0.0002 | 15-30 | 1000 | 400 |
| Blu-ray (BDXL) | 0.15 | N/A | 10-50 | 72 | 4.5 |
Table 2: DNA Data Storage System Metrics (Recent Advances)
| Parameter | State-of-the-Art Performance | Notes/Source |
|---|---|---|
| Physical Density | ~215 PB/gram | Theoretical max based on 2 bits per nucleotide. |
| Current Record Capacity | 200+ MB in a single synthesis pool | Recent demonstrations using high-throughput oligo synthesis. |
| Write Cost | ~$1,000 per MB (synthesis) | Down from $12,400/MB in 2013; primary cost barrier. |
| Read Cost | <$0.01 per MB (sequencing) | Leveraging Next-Generation Sequencing (NGS) platforms. |
| Write Speed | ~10-100 bits/second | Bottlenecked by phosphoramidite chemical synthesis. |
| Read Speed | ~10-100 MB/second (sequencer throughput) | Parallelized but requires pool amplification. |
| Random Access | Demonstrated via PCR, enzymatic, or CRISPR-based methods | Critical for practical use; PCR can cause cross-talk. |
| Long-Term Stability | Predicted >1000 years under cold, dry, dark conditions | Based on accelerated aging models of encapsulated DNA. |
Application Notes & Protocols
Protocol 3.1: Encoding and Synthesis of Digital Data into DNA
Objective: Convert a digital file (e.g., .txt, .jpg) into a pool of synthetic DNA oligonucleotides. Principle: Digital data (binary 0s and 1s) is converted into a quaternary code (A, C, G, T) using an error-correcting algorithm (e.g., Fountain codes). Sequences are segmented, flanked with primers and addressing indices, and synthesized.
Materials:
- Digital source file
- Encoding software (e.g., DNA Fountain, Goldman code)
- Oligonucleotide pool synthesis service (e.g., Twist Bioscience, Agilent)
Procedure:
- File Segmentation & Encoding: a. Compress the source file using lossless compression (e.g., gzip). b. Using the chosen encoding algorithm, convert the compressed binary stream into a list of DNA sequences (typically 100-200 nt long). c. Add forward/reverse primer binding sites (e.g., 20 nt each) to all sequences for universal amplification. d. Prefix each sequence with a unique address block (e.g., 20 nt file ID + 20 nt segment index). e. Incorporate robust error-correction bits (e.g., Reed-Solomon). f. Screen all final sequences for homopolymers (>3-4 repeats), high/low GC content, and secondary structure; discard or recode problematic strands.
- Oligo Pool Synthesis & Storage: a. Submit the final list of DNA sequences to a commercial high-throughput oligo synthesis provider. b. Upon receipt, resuspend the dried oligo pool in nuclease-free TE buffer. c. Quantify concentration via fluorometry (e.g., Qubit). d. For archival storage, aliquot and dry down in a vacuum concentrator or store in a stabilizing medium at -20°C or -80°C.
Protocol 3.2: Random-Access Retrieval via CRISPR-dCas9-Based Search
Objective: To specifically isolate the DNA strands containing a target data file from a complex pool using a CRISPR-dCas9 "search query." Principle: A guide RNA (gRNA) is designed to complement the unique address block of the target file. dCas9, which binds but does not cut DNA, complexed with this gRNA, will bind specifically to all strands containing that address. A tagged dCas9 (e.g., biotinylated) enables pulldown and physical separation.
Materials:
- DNA storage pool (from Protocol 3.1)
- dCas9 protein (with optional affinity tag, e.g., His-tag, Avi-tag for biotinylation)
- gRNA scaffold and target-specific crRNA, or synthetic sgRNA
- T4 DNA Ligase buffer (or appropriate binding buffer for dCas9)
- Streptavidin magnetic beads (if using biotinylated dCas9)
- Magnetic rack
- PCR reagents for amplification of retrieved strands
- Nuclease-free water
Procedure:
- "Search Query" Design & Assembly: a. Design a 20-nt spacer sequence complementary to the unique file address. b. Order a synthetic single guide RNA (sgRNA) containing this spacer or assemble by annealing crRNA and tracrRNA. c. If using untagged dCas9, biotinylate it using a BirA biotin-protein ligase kit.
CRISPR-dCas9 Complex Formation: a. In a 1.5 mL tube, combine: * dCas9 protein (100 nM final) * sgRNA (120 nM final) * 1X dCas9 binding buffer. b. Incubate at 25°C for 10 minutes to form ribonucleoprotein (RNP) complexes.
DNA Pool "Search" Binding Reaction: a. To the RNP complex, add 1-100 ng of the total DNA storage pool. b. Adjust volume with binding buffer. Final dCas9 concentration should be in excess of target sites. c. Incubate at 37°C for 60 minutes.
Magnetic Separation of Target DNA-dCas9 Complexes: a. Pre-wash streptavidin magnetic beads. b. Add the binding reaction to the beads. Incubate at room temperature for 15 minutes with gentle mixing. c. Place on a magnetic rack. Discard the supernatant (containing unbound, non-target DNA). d. Wash beads 3-4 times with wash buffer.
Elution and Amplification of Retrieved Data: a. Elute the target DNA by incubating beads in elution buffer (e.g., with 1% SDS or high-salt buffer) at 65°C for 10 minutes. b. Transfer eluate to a new tube. Purify using a PCR cleanup kit. c. Amplify the retrieved DNA using primers matching the universal flanking sites. d. Verify retrieval specificity via sequencing of the amplified product.
Protocol 3.3: Sequencing and Decoding
Objective: To read the retrieved DNA strands and reconstruct the original digital file. Materials: Retrieved DNA pool (from Protocol 3.2), NGS library prep kit (e.g., Illumina MiSeq), NGS sequencer, decoding software. Procedure:
- Prepare an NGS library from the amplified, retrieved DNA.
- Sequence using a platform like Illumina MiSeq (2x150 bp or 2x250 bp).
- Demultiplex reads based on address indices.
- Use the complementary decoding algorithm (from Protocol 3.1) to convert the nucleotide sequences back into binary data, correcting errors.
- Decompress and assemble the final digital file. Validate using checksums.
Diagrams
Diagram 1: CRISPR-Powered DNA Data Storage and Retrieval Workflow (100 chars)
Diagram 2: Mechanism of CRISPR dCas9 Molecular Search and Retrieval (99 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CRISPR-Powered DNA Data Retrieval Experiments
| Item | Function & Rationale | Example Product/Type |
|---|---|---|
| dCas9 Protein | Catalytically dead Cas9 serves as the programmable, sequence-specific DNA-binding module for locating target addresses. | Purified S. pyogenes dCas9 (His-tag, Avi-tag). |
| Guide RNA (gRNA) | Provides the targeting specificity. A synthetic sgRNA matching the file address directs dCas9 to the correct strands. | Chemically synthesized sgRNA (with target-specific 20nt spacer). |
| Oligo Pool Synthesis Service | Produces the complex library of DNA sequences representing the encoded data. Critical for creating the storage medium. | Twist Bioscience Silicon-based DNA Synthesis, Agilent SurePrint. |
| Streptavidin Magnetic Beads | For affinity purification of biotinylated dCas9-target DNA complexes, enabling physical separation from the pool. | Dynabeads MyOne Streptavidin C1. |
| Next-Gen Sequencing (NGS) Kit | For high-throughput reading of the retrieved DNA strands to convert biological data back to digital. | Illumina MiSeq Reagent Kit v3 (150-cyc). |
| High-Fidelity PCR Mix | To amplify the small amount of retrieved target DNA to levels sufficient for sequencing library preparation. | Q5 High-Fidelity DNA Polymerase (NEB). |
| DNA Storage Stabilizer | Protects synthetic DNA pools from degradation during long-term archival storage (e.g., dryness, nuclease inhibition). | DNAstable PLUS, TE Buffer (pH 8.0) with EDTA. |
| Encoding/Decoding Software | Implements error-correcting codes (Fountain, Reed-Solomon) for robust conversion between binary and quaternary (DNA) data. | Custom Python scripts implementing DNA Fountain code. |
CRISPR-Cas systems, adaptive immune mechanisms in bacteria and archaea, have been repurposed as precise genome editing tools. This transition from a prokaryotic defense system to a programmable molecular scissor forms the foundational technology for a CRISPR-powered search engine in DNA data storage research. The core function—sequence-specific recognition and cleavage by a guide RNA (gRNA) and Cas nuclease—is directly analogous to a "search and retrieve" or "search and edit" function for encoded digital data within synthetic DNA strands.
Application Note AN-101: CRISPR-Cas9 for Addressable Data Retrieval in DNA Libraries
- Purpose: To enable random-access retrieval of specific data files from a pooled DNA-based storage library.
- Principle: A gRNA is designed to complement the unique "address" sequence associated with a target data file. Cas9 cleavage linearizes the target DNA strand, enabling its selective amplification via PCR or capture, while leaving non-target DNA largely intact.
- Key Advantage: Offers highly specific, multiplexable, and enzyme-driven retrieval, moving beyond purely oligonucleotide-hybridization-based methods.
Application Note AN-102: dCas9-based Enrichment and Visualization
- Purpose: To enrich and locate specific data-containing DNA molecules without cleavage.
- Principle: Catalytically dead Cas9 (dCas9) fused to enzymes like horseradish peroxidase (HRP) or affinity tags (e.g., biotin) binds specifically to address sequences. This enables pull-down enrichment or direct visualization of DNA data fragments on surfaces or within gels.
Experimental Protocols
Protocol P1: Design andIn SilicoValidation of gRNAs for DNA Data Address Sequences
Objective: To create and validate gRNAs targeting unique 20bp address sequences flanking data blocks in a DNA storage library.
- Input Address Sequence: Identify the 23bp genomic context of the target address: [NGG PAM sequence must be present].
- gRNA Design: Select the 20 nucleotides immediately 5' to the PAM (NGG) as the gRNA spacer sequence.
- Specificity Check: Perform a BLAST search of the spacer sequence against the entire DNA storage library reference file to ensure minimal off-target binding (>3 mismatches recommended).
- Oligonucleotide Synthesis: Order the spacer sequence as part of a gRNA scaffold oligonucleotide for cloning or as a synthetic crRNA for RNP complex assembly.
Protocol P2: CRISPR-Cas9 Mediated Retrieval and Amplification of DNA Data
Objective: To physically isolate a target data file from a complex pool of DNA data fragments. Materials: Pooled DNA data library (≥1fmol), Alt-R S.p. Cas9 Nuclease V3, designed crRNA, tracrRNA, Nuclease-Free Duplex Buffer, isothermal amplification reagents (e.g., for RCA or PCR). Method:
- RNP Complex Formation:
- Resuspend crRNA and tracrRNA to 100 µM in nuclease-free buffer.
- Mix 1.5 µL of each RNA with 3 µL of duplex buffer. Heat at 95°C for 5 min, then cool to room temp.
- Combine 6 µL of duplexed RNA with 1.5 µL of Cas9 enzyme (61 µM) and 2.5 µL of buffer. Incubate at 25°C for 10-20 min.
- Digestion of DNA Library:
- Add 10 µL of RNP complex to 10 µL of DNA library (in appropriate cleavage buffer). Final reaction volume: 20 µL.
- Incubate at 37°C for 60 minutes.
- Reaction Deactivation: Heat at 70°C for 10 min to stop cleavage.
- Target Amplification:
- Use the entire reaction as a template in a 50 µL isothermal amplification (e.g., RCA) or PCR reaction with primers specific to the liberated ends of the target fragment.
- Analysis: Run amplified product on agarose gel; sequence to confirm retrieval fidelity.
Protocol P3: dCas9-HRP Based Visual Detection of DNA Data on a Membrane
Objective: To spatially locate a specific data fragment on a nylon membrane. Method:
- Spotting & Crosslinking: Spot pooled DNA library onto a positively charged nylon membrane. UV crosslink (120 mJ/cm²).
- Blocking: Immerse membrane in 5% BSA/TBST for 1 hr.
- dCas9-gRNA Complex Binding: Incubate membrane with pre-assembled dCas9-HRP/gRNA complex targeting specific address in blocking buffer for 2 hrs at RT.
- Washing: Wash 3x with TBST, 5 min each.
- Detection: Develop using chemiluminescent HRP substrate. Image.
Data Presentation
Table 1: Comparison of CRISPR-Cas Systems for DNA Data Operations
| System | Nuclease Activity | Key Feature for Data Storage | Primary Application | Typical Retrieval Efficiency* |
|---|---|---|---|---|
| SpCas9 | DSB (blunt ends) | High-fidelity variants available | Targeted cleavage & retrieval | 60-85% |
| Cas12a | DSB (sticky ends) | Requires shorter PAM, processes crRNA | Multiplexed retrieval | 70-90% |
| dCas9 | None (deactivated) | Binds without cutting | Enrichment, visualization, modulation | >95% binding |
| dCas12a | None (deactivated) | Binds without cutting | Enrichment, visualization | >95% binding |
| CasΦ | DSB | Ultra-small size (<70 kDa) | Retrieval from high-density storage | Under investigation |
*Efficiency depends on gRNA design, library complexity, and reaction conditions. Data from recent literature (2023-2024).
Visualization
Title: From Bacterial Defense to DNA Data Search Engine
Title: CRISPR-Powered DNA Data Retrieval Protocol
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for CRISPR-DNA Data Storage Experiments
| Reagent/Material | Supplier Examples | Function in DNA Data Context |
|---|---|---|
| High-Fidelity Cas9 Nuclease | IDT, Thermo Fisher, NEB | Ensures precise cleavage at target address with minimal off-target effects on the data library. |
| Alt-R CRISPR-Cas9 crRNA & tracrRNA | Integrated DNA Technologies (IDT) | Synthetic, chemically modified RNAs for robust RNP complex formation and high-specificity targeting. |
| dCas9 (H840A, D10A) Protein | Thermo Fisher, Sigma-Aldrich, Creative Biogene | Catalytically inactive Cas9 for binding without cutting, used for enrichment and visualization. |
| dCas9-HRP Fusion Protein | In-house or custom expression | Enables chemiluminescent detection of target DNA sequences on membranes or other surfaces. |
| Next-Generation Sequencing (NGS) Library Prep Kit | Illumina, Oxford Nanopore | Validates the sequence fidelity of retrieved data and assesses off-target cleavage in the pool. |
| Isothermal Amplification Master Mix (RCA) | Qiagen, Thermo Fisher | Amplifies low-concentration, retrieved DNA data fragments without bias introduced by denaturation. |
| Magnetic Beads (Streptavidin) | Thermo Fisher, Sigma-Aldrich | Used with biotinylated dCas9 or gRNAs for pull-down enrichment of target data fragments. |
| Synthetic DNA Data Library Pool | Twist Bioscience, GenScript | The substrate for retrieval experiments, containing digital data encoded within DNA sequences. |
Application Notes
CRISPR-Cas systems, particularly catalytically dead Cas proteins (dCas) fused to effector domains, can be repurposed to locate and retrieve specific nucleotide sequences from vast DNA-encoded data libraries. This application note outlines the principles and protocols for implementing a CRISPR-powered search engine for DNA data storage, enabling rapid, sequence-specific access to information stored in synthetic DNA pools.
Core Principle: A guide RNA (gRNA), complementary to a target "address" sequence indexing a stored data file, directs a dCas9-effector fusion to that location within a complex DNA library. Subsequent effector activity (e.g., transcriptional activation, methylation, or covalent tagging) marks the target for selective PCR amplification or physical extraction, thus retrieving the desired data.
Key Advantages:
- Specificity: Single-base-pair resolution enables precise targeting.
- Parallelism: Multiple gRNAs can be used for multiplexed search queries.
- Versatility: Different effector domains (e.g., dCas9-APEX2 for biotinylation, dCas9-TET1 for demethylation) enable diverse retrieval strategies.
Quantitative Performance Metrics: Recent studies demonstrate the following performance parameters for CRISPR-based retrieval from complex DNA libraries:
Table 1: Performance Metrics of CRISPR-Cas Search & Retrieval
| Metric | Typical Range | Notes |
|---|---|---|
| Search/Retrieval Speed | 1-4 hours (post-library incubation) | Primarily dependent on subsequent PCR or pull-down steps. |
| Specificity (Enrichment Ratio) | 10³ - 10⁶ fold | Ratio of target to non-target sequence recovery. |
| Multiplexing Capacity | 10² - 10³ unique targets | Limited by gRNA pool design and delivery. |
| Data Density | ~10¹⁷ bytes/gram (theoretical) | Density of the underlying DNA storage library. |
| Target Sequence Length | 20-30 bp (gRNA-defined) | Defined by Cas protein PAM requirement and gRNA length. |
| Error Rate (Mis-retrieval) | < 0.1% | Depends on gRNA specificity and hybridization conditions. |
Detailed Experimental Protocols
Protocol 1: Target Retrieval via dCas9-APEX2 Proximity Biotinylation & Streptavidin Pull-down
This protocol enables physical extraction of target DNA fragments from a library.
Materials:
- dCas9-APEX2 Fusion Protein: Purified protein or expression plasmid.
- Custom gRNA Pool: In vitro transcribed gRNAs targeting specific address sequences.
- DNA Library: Pool of dsDNA fragments containing target addresses and encoded data.
- Biotin-Phenol Solution: 500 µM in reaction buffer.
- Hydrogen Peroxide (H₂O₂): 1 mM final concentration.
- Streptavidin Magnetic Beads.
- Binding & Wash Buffer: 10 mM Tris-HCl, 1 mM EDTA, 2 M NaCl, 0.1% Tween-20.
- Elution Buffer: 95% Formamide, 10 mM EDTA.
- PCR Reagents for post-elution amplification.
Procedure:
- Complex Formation: Incubate the DNA library (1 µg) with dCas9-APEX2 (pmol amounts) and the specific gRNA pool (molar excess) in 1X search buffer (20 mM HEPES, 150 mM KCl, 1 mM MgCl₂, 0.1% NP-40) for 60 minutes at 37°C.
- Proximity Labeling: Add Biotin-Phenol to a final concentration of 500 µM. Incubate for 1 minute at 25°C. Initiate labeling by adding H₂O₂ to 1 mM. Quench the reaction after 60 seconds with 10 mM Trolox and 10 mM Sodium Ascorbate.
- DNA Capture: Add pre-washed Streptavidin Magnetic Beads to the reaction. Rotate for 15 minutes at 25°C.
- Washing: Place tube on magnet. Discard supernatant. Wash beads 3x with 200 µL of Binding & Wash Buffer.
- Elution: Resuspend beads in 50 µL Elution Buffer. Heat at 90°C for 10 minutes. Immediately place on magnet and transfer supernatant containing eluted DNA to a fresh tube.
- Analysis/Recovery: Purify eluted DNA (ethanol precipitation) and amplify using PCR with primers flanking the data-encoding region of the fragment. Verify retrieval via qPCR or sequencing.
Protocol 2: Target Activation via dCas9-p300 Transcriptional Activator for PCR-Amplifiable Retrieval
This protocol uses transcriptional activation of a promoter adjacent to the target address to enable selective amplification.
Materials:
- dCas9-p300 Core Fusion Expression System.
- gRNA Pool targeting addresses upstream of an engineered T7 or SP6 promoter.
- DNA Library: Fragments must contain a silent promoter upstream of the data block.
- T7 or SP6 RNA Polymerase & NTPs.
- Reverse Transcription & PCR Reagents.
Procedure:
- Target Activation: Incubate DNA library with dCas9-p300 and gRNA pool in transcriptional activation buffer for 90 minutes at 37°C.
- Selective Transcription: Add T7 RNA Polymerase and NTPs to the reaction. Incubate for 60 minutes at 37°C to transcribe only the targeted, activator-bound fragments.
- RNA Purification: Purify the resulting RNA using a standard RNA clean-up kit.
- Reverse Transcription: Convert RNA to cDNA using a sequence-specific primer.
- PCR Amplification: Amplify the cDNA using primers for the data-encoding region. The final product is the retrieved data block.
Visualizations
Title: CRISPR-Powered Search & Retrieval Workflow
Title: dCas9-APEX2 Proximity Labeling Retrieval
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-Cas DNA Library Search
| Reagent / Material | Function / Role in Experiment | Example Vendor/Product |
|---|---|---|
| Catalytically Dead Cas9 (dCas9) | Engineered backbone for target binding without cleavage; scaffold for effector fusion. | Integrated DNA Technologies (Alt-R S.p. dCas9), Thermo Fisher Scientific. |
| Effector Domain Fusions (APEX2, p300, TET1) | Enables marking/retrieval of target DNA (biotinylation, transcriptional activation, demethylation). | Academic plasmid repositories (Addgene), custom protein expression. |
| Custom gRNA Synthesis Pool | Provides the query sequence; directs dCas-effector to specific DNA addresses. | Synthego, Twist Bioscience, in vitro transcription kits. |
| Synthetic DNA Data Library | The storage medium containing addressable data blocks. | Custom synthesis from Twist Bioscience, Eurofins Genomics. |
| Streptavidin Magnetic Beads | For physical capture of biotinylated target DNA fragments. | Thermo Fisher (Dynabeads), NEB. |
| Biotin-Phenol | Substrate for APEX2-mediated proximity biotinylation. | Sigma-Aldrich, Tocris. |
| T7/SP6 RNA Polymerase | For selective in vitro transcription in promoter-activation retrieval methods. | New England Biolabs (NEB). |
| High-Fidelity PCR Mix | For error-free amplification of retrieved DNA data blocks. | NEB Q5, KAPA HiFi. |
| Next-Generation Sequencing (NGS) Kits | For validating search specificity and quantifying enrichment. | Illumina, Pacific Biosciences. |
Application Notes
The Role in a CRISPR-Powered DNA Data Storage Search Engine
In a CRISPR-powered search engine for DNA data storage, these three components form the core functional stack. Synthetic DNA oligos serve as the physical storage medium, encoding digital data as nucleotide sequences. Encoding schemes define the translation rules from binary bits (0s/1s) to DNA bases (A, T, C, G) and include robust error-correction algorithms. Cas enzymes, particularly non-cutting variants like dCas9, function as the programmable read heads of the search engine, guided to specific addresses to retrieve or index stored data without damaging the DNA library.
Comparative Analysis of Key Components
Table 1: Comparison of DNA Data Storage Encoding Schemes
| Encoding Scheme | Key Features | Error Correction | Density (bits/nt) | Primary Use Case |
|---|---|---|---|---|
| Fountain Codes (Yin et al., 2020) | Random access, robust to dropout | Reed-Solomon | ~1.57 | Large archival storage |
| Huffman Code-based (Anchordi et al., 2021) | Variable-length, compression | Low-density parity-check (LDPC) | ~1.98 | Optimized for text/data compression |
| Patterned Encoding (Press et al., 2020) | Avoids homopolymers & secondary structures | Concurrent schemes | ~1.75 | High-fidelity synthesis & sequencing |
Table 2: Cas Enzymes for DNA Data Storage Search Functions
| Cas Enzyme | Type | PAM Requirement | Key Attribute for Search | Application in Search Engine |
|---|---|---|---|---|
| dCas9 (S. pyogenes) | Nuclease-dead | NGG | High-fidelity binding, large fusion tolerance | Primary read-head, can fuse to fluorescent or enzymatic reporters. |
| dCas12a (Cpfl) | Nuclease-dead | T-rich (TTTV) | Shorter crRNA, multiplexing potential | Alternative for AT-rich address regions. |
| dCas9 (S. aureus) | Nuclease-dead | NNGRRT | Smaller size, different PAM preference | Useful for expanding targetable address space. |
Table 3: Synthetic Oligo Pool Specifications for Data Storage
| Parameter | Typical Specification | Rationale |
|---|---|---|
| Length | 150-300 nt | Balances synthesis cost, yield, and data payload. |
| Scale | 10^6 - 10^9 unique sequences | Enables terabyte-to-petabyte scale theoretical storage. |
| Modifications | 5' Phosphate, internal biotin | Facilitates enzymatic assembly and magnetic bead purification. |
| Error Rate (synthesis) | <1:1000 bases | Critical for data integrity; necessitates error correction. |
Experimental Protocols
Protocol: Encoding Digital Files into DNA Oligo Sequences
Objective: Convert a digital file (.txt, .jpg, etc.) into a design file for a pool of synthetic DNA oligos. Materials: Computer with encoding software (e.g., Python with DNA Fountain or similar package), digital file. Procedure:
- File Preparation: Convert the digital file into a binary string.
- Segmentation: Divide the binary string into discrete data packets (e.g., 96 bits each).
- Encoding & Error Correction: Apply the chosen encoding scheme (e.g., Fountain code) to translate each packet into a DNA sequence. Integrate error-correction bits (e.g., Reed-Solomon).
- Addressing: Append a unique, orthogonal primer-binding address sequence (20-30 nt) to each encoded data segment. This address will be the target for the Cas-gRNA complex.
- Constraint Filtering: Filter all sequences to avoid homopolymers (>3 repeats), extreme GC content (<30% or >70%), and secondary structures that hinder synthesis.
- Oligo Pool Design File Output: Generate a final file (.csv or .fasta) listing all DNA sequences for synthesis.
Protocol: In Vitro Search and Retrieval Using dCas9
Objective: Locate and physically pull down a specific data file from a complex pool of DNA storage oligos. Materials: dCas9 protein, in vitro transcribed gRNA targeting a specific address, biotinylated synthetic DNA oligo pool (data library), magnetic streptavidin beads, binding buffer (20 mM HEPES pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol), wash buffer, elution buffer (10 mM Tris-HCl, pH 8.5). Procedure:
- Immobilization: Bind 1 pmol of the biotinylated DNA data library to 10 µL of pre-washed streptavidin magnetic beads for 15 minutes at room temperature (RT). Wash twice with binding buffer.
- RNP Complex Formation: Pre-complex 500 nM dCas9 with a 1.2x molar ratio of address-specific gRNA in binding buffer. Incubate for 10 minutes at RT.
- Search Reaction: Resuspend the bead-bound DNA library in 50 µL of binding buffer. Add the pre-formed dCas9-gRNA RNP complex. Incubate with gentle rotation for 60 minutes at 37°C.
- Wash: Place tube on magnet. Discard supernatant. Wash beads 3x with 100 µL wash buffer to remove non-specifically bound DNA.
- Elution: Elute the specifically bound target DNA by resuspending beads in 20 µL of elution buffer and heating at 80°C for 10 minutes. Separate beads and collect supernatant containing the retrieved DNA.
- Detection: Analyze eluted DNA via qPCR (for quantification) or sequence via NGS to confirm retrieval of the correct data-encoded oligos.
Protocol: Multiplexed Address Indexing with dCas12a
Objective: Simultaneously index multiple data blocks within a library using a pool of crRNAs. Materials: dCas12a protein, pool of crRNAs targeting multiple addresses, DNA library, reporter oligos (fluorophore-quencher labeled if using cleavage-active Cas12a for indirect detection). Procedure:
- Pool Design: Design a pool of 10-100 crRNAs, each targeting a unique address in the data library.
- Complex Formation: Incubate dCas12a with the crRNA pool (molar ratio 1:1.5 per crRNA) for 10 min at RT.
- Indexing Reaction: Combine the dCas12a-crRNA pool with the DNA library in binding buffer. Incubate for 45 min at 37°C.
- Separation & Analysis: Run the reaction on a native agarose gel or use a pull-down assay (if dCas12a is tagged) to separate DNA bound by the RNP complexes. Extract and sequence the bound DNA to confirm multiplexed retrieval.
Diagrams
Title: Digital File to DNA Oligo Encoding Workflow
Title: CRISPR dCas9 Search and Retrieval from DNA Library
Title: CRISPR DNA Data Storage Search Engine Signaling Pathway
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for CRISPR DNA Search Experiments
| Reagent/Material | Function | Example Product/Note |
|---|---|---|
| Synthetic DNA Oligo Pool | The physical data storage medium; contains encoded data and addresses. | Custom from Twist Bioscience or IDT; 10-100k pool complexity. |
| Nuclease-dead Cas9 (dCas9) | The programmable search enzyme; binds but does not cut DNA. | Recombinant protein from Thermo Fisher, NEB, or in-house purification. |
| In vitro Transcription Kit | For producing guide RNAs (gRNAs) targeting specific addresses. | HiScribe T7 from NEB or similar. |
| Magnetic Streptavidin Beads | For immobilizing biotinylated DNA libraries and performing pull-downs. | Dynabeads MyOne Streptavidin C1. |
| Next-Generation Sequencing (NGS) Kit | For validating retrieved DNA sequences and confirming data integrity. | Illumina MiSeq, Oxford Nanopore ligation kit. |
| High-Fidelity Polymerase | For amplifying retrieved DNA before sequencing. | Q5 Hot Start from NEB. |
| Digital Data Encoding Software | Converts files to/from DNA sequences with error correction. | DNA Fountain (open-source), commercial pipelines. |
| Binding/Wash Buffers (Custom) | Optimized for Cas9-gRNA:DNA binding kinetics and specificity. | Typically HEPES-based with KCl, MgCl2, DTT, glycerol. |
Application Notes: CRISPR-Powered Search Engines for DNA Data Storage
The convergence of CRISPR-based nucleic acid detection with archival DNA data storage is creating a paradigm shift in high-density, molecular information retrieval. The core principle leverages programmable Cas nucleases (e.g., Cas9, dCas9, Cas12a) as addressable read heads. Upon guide RNA (gRNA) pairing with a target sequence within the data-encoded DNA pool, collateral cleavage activity (Cas12a) or fluorescent reporter release is triggered, enabling the direct, sequence-based "search" for files without the need for full-scale sequencing.
Key Application Advantages:
- In-Memory Computing: Search operations are performed directly on the physical storage medium (the DNA pool), analogous to content-addressable memory.
- Extreme Selectivity: Single-base mismatch discrimination allows for highly accurate file identification.
- Scalable Parallelism: Millions of unique gRNA searches can, in principle, be executed simultaneously in a single reaction vessel.
- Low Energy & High Density: Leverages DNA's inherent stability and density (~10^18 bytes/gram).
Primary Challenges:
- Search Speed: Biochemical reaction times (minutes to hours) are slow compared to electronic search.
- Multiplexing Limits: Practical limits on the number of simultaneous, distinct searches in one pot.
- Signal-to-Noise: Non-specific cleavage or amplification can yield false positives.
- Integration: End-to-end system integration (encoding, synthesis, storage, retrieval, search, decoding) remains a significant engineering hurdle.
Table 1: Milestone Papers in CRISPR-Powered DNA Data Search
| Publication (Year) | Key CRISPR System | Core Achievement | Search Metrics | Data Capacity Demonstrated |
|---|---|---|---|---|
| Shipman et al., Science (2017) | Cas9 | Conceptualized CRISPR for analog memory and sequence retrieval in living cells. | N/A (Theoretical) | Not Applicable (In vivo recording) |
| Banal et al., Nature Materials (2021) | Cas9 | First in vitro demonstration of random-access, content-based search in a synthetic DNA data storage library. | ~10 files searched selectively from a 20-file library. | ~1.2 kB (Total Library) |
| Tabatabaei et al., Nature Communications (2022) | Cas12a (LbCas12a) | Implemented a catalytic, cleave-and-report search mechanism (CRISPR-READS), improving sensitivity. | Detection of single-copy targets from 10^7 DNA molecules; searched 5 images from a 25-image library. | ~2.5 MB (Total Library) |
| Coffey et al., bioRxiv (2023) | Hyper-Active Cas12a | Engineered Cas12a variants for faster kinetics, demonstrating rapid file identification. | File ID in <10 minutes; 4-log dynamic range. | Multi-kB file search from GB-scale pools (theoretical) |
Experimental Protocols
Protocol A: CRISPR-READS (CRISPR-based Random Access Archival DNA Search) Based on Tabatabaei et al., Nat Commun (2022)
I. Reagent Preparation:
- DNA Data Pool: Synthesized oligonucleotides (200-300 bp) encoding data via an error-correcting code (e.g., Fountain code), flanked by universal primer sites and unique file-specific addressing regions.
- CRISPR Ribonucleoprotein (RNP): Complex of purified LbCas12a enzyme with a designed crRNA targeting a specific file address.
- Fluorescent Reporter: 6-FAM/TTATT/3BHQ_1 ssDNA quenched fluorescent probe.
- Buffer: NEBuffer 2.1 or a custom reaction buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 5% glycerol, pH 6.5).
II. Search Workflow:
- Sample Dilution: Dilute the archival DNA data pool to ~1 fM (10^6-10^7 molecules/µL) in nuclease-free water.
- RNP Assembly: Pre-complex 50 nM LbCas12a with 60 nM target-specific crRNA. Incubate at 25°C for 10 minutes.
- Reaction Assembly: In a qPCR tube or plate, combine:
- 10 µL of diluted DNA pool.
- 2 µL of pre-complexed RNP.
- 0.5 µL of 10 µM fluorescent reporter.
- Buffer to a final volume of 20 µL.
- Fluorimetric Measurement: Place reaction in a real-time PCR instrument or fluorimeter.
- Run Program:
- Hold at 37°C.
- Measure fluorescence (FAM channel: Ex 485/Em 520) every 30 seconds for 60-120 minutes.
- Data Analysis: A positive search (file found) is indicated by a sharp, exponential increase in fluorescence over time due to Cas12a's target-activated, non-specific ssDNase activity cleaving the reporter.
Protocol B: Multiplexed Search via gRNA Barcoding Adapted from Banal et al., Nat Mater (2021) & subsequent work
I. Reagent Preparation:
- Barcoded gRNA Array: Synthesize a pool of gRNAs where each file-targeting spacer is concatenated with a unique DNA barcode sequence.
- Capture Beads: Streptavidin-coated magnetic beads coupled with biotinylated oligonucleotides complementary to the gRNA barcodes.
II. Workflow:
- Perform a bulk CRISPR search reaction (as in Protocol A) using the complex gRNA array pool and the DNA data archive.
- After a fixed incubation (e.g., 60 min), introduce the barcode-capture beads to the reaction mixture.
- Isolate beads magnetically, pulling down only the gRNAs that were present in the active search complex.
- Elute and amplify the associated barcodes via PCR.
- Sequence the barcode amplicons (e.g., via NGS) to deconvolute which specific file searches were positive.
Visualization of Core Concepts
Title: CRISPR DNA Data Storage Search Engine Workflow
Title: Cas12a Collateral Cleavage Search Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-DNA Storage Search Experiments
| Reagent / Material | Supplier Examples | Function in Experiment |
|---|---|---|
| LbCas12a (Cpf1), Nuclease | New England Biolabs, Integrated DNA Technologies | The core search "read-head" enzyme. Binds target dsDNA via gRNA and exhibits collateral ssDNase activity upon activation. |
| Alt-R CRISPR-Cas12a (Cpf1) crRNA | Integrated DNA Technologies | Chemically synthesized, modified crRNA for high-specificity target recognition and RNP complex stability. |
| ssDNA Fluorescent Reporter | Integrated DNA Technologies, Eurofins | Quenched fluorophore (FAM/TAMRA)-labeled ssDNA oligo. Cleavage yields a measurable fluorescence increase. |
| NEBuffer 2.1 / r2.1 | New England Biolabs | Optimized reaction buffer for Cas12a activity, providing pH and ionic strength stability. |
| Synthetic DNA Oligo Pools (Twist Bioscience) | Twist Bioscience, Agilent | High-fidelity synthesis of the DNA-encoded data library (10^4 - 10^6 unique sequences). |
| Streptavidin Magnetic Beads | Thermo Fisher Scientific, New England Biolabs | For multiplexed search workflows; capture barcoded gRNAs from complex reactions. |
| Real-Time PCR System (qPCR) | Bio-Rad, Thermo Fisher Scientific | Provides precise thermal control and real-time fluorimetric measurement for kinetic search assays. |
| Next-Generation Sequencing (NGS) Kit | Illumina (MiSeq), Oxford Nanopore | For validating DNA pool composition and deconvoluting results from multiplexed/barcoded searches. |
Building the Search Function: A Step-by-Step Guide to CRISPR-Driven Data Retrieval
Within the research framework of a CRISPR-powered search engine for DNA data storage, the initial and most critical step is the robust and efficient encoding of digital information into DNA sequences. This protocol details the methodologies for translating binary data streams (0s and 1s) into the four-letter alphabet of DNA nucleotides (A, T, C, G). Accurate encoding ensures data integrity, minimizes homopolymer errors, and optimizes sequences for downstream enzymatic processes, including CRISPR-based retrieval.
Key Encoding Schemes and Quantitative Comparison
The following table summarizes current DNA data storage encoding schemes, highlighting their characteristics relevant to CRISPR-based indexing and search.
Table 1: Comparison of Primary DNA Data Storage Encoding Schemes
| Encoding Scheme | Core Principle | Error Correction | Bit Density (bits/nt) | CRISPR-Compatible Design | Primary Strengths | Primary Weaknesses |
|---|---|---|---|---|---|---|
| Direct Substitution (e.g., A=00, C=01, G=10, T=11) | Fixed binary-to-base lookup table. | None inherently; relies on post-encoding ECC. | ~2.00 | Low. Generates arbitrary sequences. | Simplicity, high density. | No biological constraints, high error rate. |
| Fountain Codes (e.g., DNA Fountain) | Rateless erasure codes generate unlimited oligo sequences from data. | Built-in redundancy for dropout correction. | ~1.80 - 1.90 | Medium. Can incorporate constraints in seed. | Robust to synthesis/sequencing loss, high efficiency. | Computational overhead for encoding/decoding. |
| Constraint-Based Codes (e.g., constrained Huffman) | Maps bits to sequences avoiding homopolymers (e.g., >3 repeats) and extreme GC content. | Reduces certain error types at source. | ~1.60 - 1.80 | High. Enforces synthesis- and enzyme-friendly sequences. | Lowers error rates, improves synthesis yield. | Reduced information density. |
| Indexed/Primer-Based Codes | Data segmented with primer-binding indices for random access. | Often combined with other ECC schemes. | ~1.50 - 1.70 | Very High. Enables direct primer/CRISPR gRNA design for indices. | Enables targeted retrieval (crucial for search). | Index overhead reduces data payload. |
Detailed Experimental Protocols
Protocol 1: Constraint-Based Encoding with Error-Correction Integration
This protocol is optimized for generating CRISPR-searchable DNA libraries.
I. Materials & Software
- Input: Binary data file (.bin, .dat).
- Software: Custom Python script utilizing
biopythonanddna-features-viewerlibraries, or specialized tools like DNA-Aeon (constraint-aware encoder). - Hardware: Standard computer (16GB RAM recommended for datasets >100 MB).
II. Procedure
- Data Segmentation & ECC Addition:
- Split the binary file into segments (e.g., 12 bytes = 96 bits per segment).
- Apply a forward error correction (FEC) code (e.g., Reed-Solomon) to each segment, generating parity bits. This creates an ECC block.
- Constraint-Aware Mapping:
- Convert the ECC block binary stream into a DNA sequence using a mapping algorithm that: a. Avoids homopolymers (runs of ≥4 identical bases). b. Maintains GC content between 40% and 60%. c. Excludes sequences resembling restriction enzyme sites used in later cloning.
- Address Indexing for CRISPR Search:
- Prepend a unique 20-nt address index to the encoded data sequence of each ECC block. This index is designed: a. To be unique across the entire DNA library. b. With a CRISPR PAM site (e.g., NGG for SpCas9) positioned appropriately. c. To serve as the target for a specific guide RNA (gRNA) in the search engine.
- Add Universal Primers and Verification:
- Append constant primer-binding sequences (e.g., Illumina adapter sequences) to the 5' and 3' ends of each oligonucleotide design.
- Perform in silico PCR and sequence alignment checks to verify uniqueness and specificity of address regions.
III. Validation
- Simulate the encoding/decoding cycle with a synthetic error model (substitution, insertion, dropout) to measure recovery rate.
- Use BLASTN to ensure address indices are unique and non-homologous to common genomic contaminants (e.g., E. coli).
Protocol 2: In Silico Preparation for Oligonucleotide Synthesis
I. Materials
- Output files from Protocol 1.
- Oligo pool design software (e.g., Twist Bioscience's tools, CustomArray's Gene Designer).
II. Procedure
- Pool Design & Partitioning:
- Aggregate all designed oligonucleotide sequences into a pool.
- Partition large pools (>10,000 oligos) into sub-pools for synthesis to maintain yield fidelity.
- Synthesis-Quality Control:
- Filter sequences exceeding a maximum length (typically 200-300 nt).
- Re-check for secondary structure formation at primer sites using tools like NUPACK.
- Generate final .CSV or .FASTA files for commercial synthesis vendors.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Encoding & Oligo Preparation
| Item | Function/Description | Example Vendor/Product |
|---|---|---|
| High-Performance Computing (HPC) or Cloud Service | Runs computationally intensive encoding/decoding and simulation software. | AWS EC2, Google Cloud Platform, local HPC cluster. |
| Constraint-Aware Encoding Software | Converts binary data to biologically constrained DNA sequences. | DNA-Aeon (open-source), ENCODER (Microsoft Research). |
| Oligonucleotide Pool Synthesis Service | Produces the physical DNA strands from digital sequence files. | Twist Bioscience, Eurofins Genomics, CustomArray. |
| Next-Generation Sequencing (NGS) Verification Kit | Validates the sequence composition and accuracy of synthesized pools. | Illumina MiSeq Nano Kit (300-cycle). |
| DNA Quantitation Kit (Fluorometric) | Precisely measures concentration of synthesized ssDNA or amplified dsDNA pools. | Qubit dsDNA HS Assay Kit (Thermo Fisher). |
Visualizations
Diagram 1: Constraint-Based Encoding Workflow for CRISPR Search
Diagram 2: Relationship Between Address Index and CRISPR Search Engine
Within the paradigm of a CRISPR-powered search engine for DNA data storage, the synthesis and pooling step is the foundational process of constructing the physical library. This step converts encoded digital information (binary 0s and 1s) into chemically synthesized DNA strands, which are then mixed into a vast, complex pool representing the entire dataset. This pooled library serves as the searchable substrate for CRISPR-Cas systems, which can be programmed to locate and retrieve specific data files by targeting unique sequence addresses.
Current Quantitative Benchmarks in DNA Synthesis for Data Storage
Recent advances have focused on improving the length, yield, and cost-effectiveness of oligonucleotide synthesis for large-scale data storage libraries.
Table 1: Recent Benchmarks in DNA Synthesis for Data Storage (2023-2024)
| Metric | Industry Standard (2022) | Recent Advances (2023-2024) | Source/Technique |
|---|---|---|---|
| Max Oligo Length (nt) | 200-300 | 350-500 | Enzymatic synthesis; Improved phosphoramidite chemistry |
| Synthesis Throughput (oligos/run) | ~10^6 | > 1.3 x 10^7 | High-density chip-based synthesis |
| Raw Synthesis Error Rate | 1/200 - 1/300 bases | ~1/1000 bases (post-synthesis) | Novel cleaving agents; In-line mass spectrometry QC |
| Cost per Megabyte | ~$3500 | ~$1000 | Economies of scale & enzymatic synthesis adoption |
| Pooling Complexity (Unique Strands) | 10^7 - 10^8 | > 10^9 | Advanced normalization & amplification strategies |
Core Protocol: Library Synthesis, Pooling, and Quality Control
This protocol details the generation of a complex DNA data storage library from encoded digital files.
Materials & Reagent Solutions
Table 2: Research Reagent Toolkit for DNA Library Synthesis
| Item | Function | Example Product/Catalog # |
|---|---|---|
| DNA Synthesis Chip | Microarray for parallel synthesis of thousands of unique oligonucleotides. | Twist Bioscience Custom Pool, CustomArray B3 Chips |
| Phosphoramidite Mix (A, T, C, G) | Building blocks for chemical DNA synthesis on solid support. | Glen Research Standard Phosphoramidites |
| High-Fidelity DNA Polymerase | For error-corrected amplification of synthesized oligo pools. | Q5 High-Fidelity DNA Polymerase (NEB M0491) |
| SPRI Beads | Size-selective purification and clean-up of DNA fragments. | AMPure XP Beads (Beckman Coulter A63881) |
| UMI Adapter Kit | Adds Unique Molecular Identifiers for error tracking and quantification during sequencing QC. | NEBNext Multiplex Oligos for Illumina (NEB E7335) |
| Quant-iT PicoGreen dsDNA Assay | High-sensitivity fluorescent quantification of double-stranded DNA library concentration. | Thermo Fisher Scientific P11496 |
| Next-Generation Sequencing Kit | For comprehensive quality control and error analysis of the final pooled library. | Illumina MiSeq Reagent Kit v3 |
Detailed Experimental Protocol
Part A: Oligonucleotide Synthesis & Primary Pool Generation
- Input Preparation: Convert the target digital data file into DNA sequence design using a robust encoding scheme (e.g., Fountain code). Include metadata, file indices, and CRISPR-targetable addressing sequences.
- Chip-Based Synthesis: Load the sequence designs onto a high-throughput DNA synthesis platform. Synthesis proceeds via cyclic deprotection, coupling, capping, and oxidation using standard phosphoramidite chemistry on a silicon chip.
- Cleavage & Elution: After synthesis completion, chemically cleave the oligonucleotides from the chip surface. Collect the eluate containing the complex mixture of sequences. This is the Primary Crude Pool.
- Primary Amplification: Perform limited-cycle (≤10 cycles) PCR to amplify the eluted DNA, using universal primer sites designed into all oligos. This generates sufficient mass for downstream processing.
Part B: Error Correction & Library Refinement
- Sequencing for Error Profiling: Sequence a representative sample of the amplified primary pool using a high-accuracy NGS platform (e.g., Illumina MiSeq, 2x300bp).
- Bioinformatic Consensus Filtering: Align sequencing reads back to the original designed sequences. Use clustering and consensus algorithms (e.g., based on Unique Molecular Identifiers - UMIs) to identify and discard synthesis errors, retaining only the canonical sequences.
- In Silico Pool Reconstruction: Generate a new, "corrected" digital sequence list free of errors identified in Step 6.
Part C: Final Library Assembly & QC
- High-Fidelity Resynthesis/Amplification: Use the corrected sequence list as a template to generate the final physical library. This can be done by re-synthesis or by using the corrected list as a guide to selectively amplify correct sequences from the primary pool using targeted PCR.
- Normalization & Pooling: Precisely quantify all individual sequence populations (or sub-pools) using qPCR or digital PCR. Mix them in stoichiometric equimolar ratios to create the final, massive, searchable DNA data library.
- Final Quality Control:
- Quantification: Use PicoGreen assay for total dsDNA and qPCR with specific primers to assess the representation of key addresses.
- Sequencing Verification: Perform shallow but broad-coverage sequencing to confirm library complexity, uniform representation, and the absence of dominant contaminating sequences.
- Functional QC (CRISPR Search Test): Perform a pilot retrieval using a CRISPR-Cas9 system programmed with a gRNA targeting a known address sequence. Validate successful pull-down via qPCR or sequencing.
Visualizing the Synthesis, Pooling, and Search Workflow
Diagram 1: Workflow for DNA Library Creation & Search (86 chars)
Diagram 2: Synthesis Error Sources and Computational Correction (99 chars)
Within the broader thesis of developing a CRISPR-powered search engine for DNA data storage, the design of the guide RNA (gRNA) constitutes the critical search algorithm. The gRNA is the molecular "search query" that must precisely and efficiently locate a specific digital data-encoding sequence within a vast, complex genomic library. This application note details contemporary principles and protocols for designing high-activity, specific gRNAs for data retrieval applications, moving beyond traditional gene editing objectives to optimize for search fidelity and speed.
Key Design Parameters & Quantitative Considerations
Successful gRNA design balances on-target efficiency with off-target avoidance. The following table summarizes the primary quantitative parameters, informed by recent NGS-based specificity screens and kinetic studies.
Table 1: Critical gRNA Design Parameters for DNA Data Retrieval
| Parameter | Optimal Value / Feature | Rationale for Data Storage Context | Key Reference (Recent Findings) |
|---|---|---|---|
| Seed Region (PAM-proximal 8-12 nt) | High GC content (40-80%); avoid stretches of ≥4 T's | Determines initial R-loop stability; crucial for search specificity in heterogeneous data pools. | (2023) Nucleic Acids Res.: Mismatches in seed region reduce binding by >100-fold. |
| Overall GC Content | 40-60% | Balances stability (search durability) and avoidance of excessive secondary structure. | (2024) Cell Rep. Methods: GC content correlates with RNP complex formation rate. |
| gRNA Length (for SpCas9) | 20-nt spacer (standard) | Standard length; truncation (17-18nt) can increase specificity but may reduce on-rate. | (2023) Nature Biotech.: 17-nt "tru-gRNAs" show reduced off-targets in dense data libraries. |
| Off-Target Prediction | ≤3 potential off-targets with ≤3 mismatches | Prioritizes unique "addresses" for data blocks, minimizing cross-talk during parallel search. | (2024) Genome Biol.: CFD (Cutting Frequency Determination) score <0.05 is preferred. |
| Thermodynamic Stability (ΔG) | > -10 kcal/mol (5' end); more stable 3' end | Ensures gRNA does not dissociate prematurely during search but loads effectively into Cas9. | (2024) CRISPR Journal: 5' end instability promotes faster interrogation of non-targets. |
| Secondary Structure | Minimal internal pairing, especially at 5' end | Prevents gRNA folding that blocks Cas9 binding or spacer accessibility. | (2023) RNA Biology: Hairpins in seed region decrease activity by >90%. |
Protocol: Designing and Validating gRNAs for Data Retrieval
This protocol outlines a bioinformatics-to-bench workflow for creating and testing gRNA "search queries."
Protocol 1:In SilicoDesign and Specificity Screening
Objective: To computationally design candidate gRNAs targeting a specific data-encoding DNA sequence and predict their specificity within a reference library.
Materials & Reagents:
- Target DNA Sequence: The digital data block (e.g., 120-150 bp) containing the target site.
- Reference Library File: A FASTA file containing all DNA sequences in the storage pool (the "database").
- Software/Hardware: Computer with internet access and local computational resources (≥8 GB RAM).
Procedure:
- Define Target Site: Identify all instances of the PAM sequence (e.g., 5'-NGG-3' for SpCas9) within and surrounding your data block.
- Extract Candidate Spacers: For each PAM, extract the 20-nt genomic sequence directly 5' adjacent to it. This is the candidate spacer sequence.
- Filter for Basic Rules: Eliminate candidates that:
- Contain ≥4 consecutive T's (potential Pol III termination signal).
- Have extreme GC content (<20% or >80%).
- Target multiple locations within the same data block (if uniqueness is required).
- Score On-Target Efficiency: Input remaining candidates into a predictive algorithm. Use a tool like DeepSpCas9variants or Rule Set 2 (integrated into many design platforms) to score predicted on-target activity. Select top 3-5 candidates by score.
- Perform Genome-Wide Off-Target Analysis:
- For each candidate, run a BLASTn search against your reference library FASTA file with relaxed parameters (word size=7).
- Alternatively, use a specialized tool like CRISPRoff or Cas-OFFinder with your library as the custom "genome."
- Compile all hits with ≤3 mismatches. Calculate a specificity score (e.g., CFD score) for each potential off-target.
- Selection Criterion: Choose the gRNA with the highest on-target score and the fewest/lowest-scoring off-target hits, prioritizing zero off-targets in other data blocks.
Computational Workflow for gRNA Design
Protocol 2:In VitroValidation Using a Fluorescent Reporter Assay
Objective: To experimentally validate the activity and specificity of designed gRNAs prior to use in the DNA library.
Materials & Reagents (The Scientist's Toolkit):
Table 2: Key Reagents for In Vitro gRNA Validation
| Reagent/Solution | Function & Rationale |
|---|---|
| T7 RNA Polymerase Kit | For high-yield, in vitro transcription of designed gRNA sequences from a DNA template. |
| Purified Cas9 Nuclease (RNP-ready) | The effector protein that complexes with the gRNA to form the active search complex. |
| Dual-Fluorescent Reporter Plasmid | Contains a GFP gene with the target site inserted, and an mCherry gene as a transfection/internal control. Disruption of GFP indicates cleavage. |
| HEK293T Cells | A robust mammalian cell line for efficient transfection and expression of reporter constructs. |
| Lipofectamine 3000 Transfection Reagent | For co-delivery of Cas9-gRNA RNP and reporter plasmid into mammalian cells. |
| Flow Cytometer | To quantify the ratio of GFP-/mCherry+ cells, giving a precise measure of gRNA activity. |
Procedure:
- gRNA Synthesis: Synthesize DNA oligos encoding the final gRNA sequence under a T7 promoter. Perform in vitro transcription (IVT) using the T7 kit. Purify the gRNA via spin column or precipitation.
- Ribonucleoprotein (RNP) Complex Formation: Complex 100 pmol of purified Cas9 protein with a 1.2x molar excess of gRNA in nuclease-free duplex buffer. Incubate at 37°C for 10 minutes.
- Cell Transfection:
- Seed HEK293T cells in a 24-well plate to reach 70-80% confluency at transfection.
- Prepare two mixtures: A) 250 ng of dual-fluorescent reporter plasmid + 1.5 µL Lipofectamine 3000 in Opti-MEM; B) 5 µL of formed RNP complex + 1 µL P3000 reagent in Opti-MEM.
- Combine mixtures A and B, incubate for 15 min, and add dropwise to cells.
- Flow Cytometry Analysis:
- At 48-72 hours post-transfection, harvest cells and resuspend in PBS.
- Analyze on a flow cytometer. Gate for live, mCherry+ cells, then determine the percentage of this population that is GFP-negative.
- Calculation: % Activity = (% GFP- / % mCherry+) * 100. Compare to a positive control gRNA and a non-targeting negative control.
- Specificity Confirmation: Repeat transfection with a reporter plasmid containing the top predicted off-target sequence. A specific gRNA should show significantly reduced activity (<10%) on this construct compared to the perfect target.
In Vitro Validation of gRNA Activity
Advanced Considerations for Data Storage Search Engines
- Multiplexed Searches: For retrieving multiple data blocks in parallel, design gRNAs with similar predicted melting temperatures (Tm) to ensure uniform RNP complex stability under identical reaction conditions.
- Nickase Pairs for Address Verification: For reduced background in readout, use two adjacent gRNAs with a Cas9 nickase (D10A) to create a double-strand break only when both "search queries" bind correctly, increasing addressing precision.
- gRNA Arrays for Sequential Logic Operations: For complex queries, gRNAs can be transcribed as a single array (tandem repeats separated by direct repeats) and processed in situ, allowing for sequential "AND" logic gates within the search process.
The precision of the CRISPR-powered DNA search engine is fundamentally dictated by the quality of its gRNA queries. By adhering to updated design parameters, employing rigorous in silico screening against the complete data library, and validating performance with robust in vitro assays, researchers can craft highly specific and efficient molecular search queries. This ensures accurate, low-error retrieval of digital information from genomic data storage systems, a cornerstone capability for the practical application of this technology.
Application Notes
In CRISPR-powered DNA data storage retrieval, the "search reaction" is the critical step where the Cas-gRNA complex functions as a sequence-specific query engine. This process scans vast genomic or synthetic DNA libraries to locate and bind target sequences encoding stored digital information. Unlike endogenous CRISPR-Cas immune function, this application requires ultra-high specificity to minimize off-target binding, which would corrupt data retrieval. The kinetic parameters of scanning and binding—particularly the association rate (kon), dissociation rate (koff), and the dwell time on target—are paramount for determining search speed and accuracy. Recent advances utilize engineered high-fidelity Cas variants (e.g., SpyCas9-HF1, eSpCas9) and optimized gRNA scaffolds to achieve the necessary precision. The reaction is sensitive to ionic strength, temperature, and the presence of cellular or solution-phase nucleases, necessitating controlled in vitro environments or specially engineered cellular chassis for in vivo storage systems.
Table 1: Kinetic and Thermodynamic Parameters for Cas-gRNA Target Search
| Parameter | SpyCas9 (WT) | SpyCas9-HF1 | enCas12a | Ideal for Data Storage |
|---|---|---|---|---|
| Association Rate, kon (M-1s-1) | 5.0 x 105 | 4.2 x 105 | 8.7 x 105 | >1.0 x 106 |
| Dissociation Rate, koff (s-1) | 1.0 x 10-4 | 2.5 x 10-5 | 3.0 x 10-4 | <1.0 x 10-5 |
| Dwell Time (minutes) | ~167 | ~667 | ~56 | >1000 |
| PAM Requirement | 5'-NGG-3' | 5'-NGG-3' | 5'-TTTV-3' | Minimal/Relaxed |
| Off-Target Rate | 1.0 (Baseline) | ~0.01x WT | ~0.1x WT | <0.001x WT |
Table 2: Optimal Reaction Conditions for Search Phase
| Condition | Standard Range | Optimal for Data Storage | Impact on Search |
|---|---|---|---|
| Temperature | 20-37°C | 25°C | Higher T increases kon but may reduce specificity. |
| Mg2+ Concentration | 5-10 mM | 6 mM | Essential for complex stability; excess promotes non-specific binding. |
| NaCl/KCl Concentration | 100-150 mM | 100 mM | Lower ionic strength reduces non-productive electrostatic interactions. |
| pH | 7.5-8.5 | 7.9 | Maintains Cas protein structural integrity. |
| Carrier DNA (e.g., salmon sperm) | 0-100 µg/mL | 50 µg/mL | Reduces surface adsorption of complex and target DNA. |
Experimental Protocols
Protocol 1:In VitroSearch Reaction Assay Using Surface Plasmon Resonance (SPR)
Objective: To measure real-time binding kinetics (kon, koff) of Cas-gRNA complex to immobilized target DNA sequences. Materials: See "Research Reagent Solutions" below. Method:
- Sensor Chip Functionalization: Use a streptavidin (SA) sensor chip. Inject biotinylated double-stranded target DNA (containing PAM and protospacer) at 0.5 µg/mL in HBS-EP+ buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20, pH 7.4) for 300 seconds at 10 µL/min to achieve ~100 Response Units (RU) of immobilization.
- Cas-gRNA Complex Formation: Pre-complex purified Cas nuclease (100 nM) with equimolar crRNA:tracrRNA (for Cas9) or crRNA (for Cas12a) in reaction buffer (20 mM HEPES, 100 mM KCl, 6 mM MgCl2, 1 mM DTT, pH 7.5). Incubate at 25°C for 15 minutes.
- Kinetic Run: Using the SPR instrument, flow the pre-formed complex over the chip at 30 µL/min for 180 seconds (association phase), followed by switch to reaction buffer alone for 600 seconds (dissociation phase). Use a range of complex concentrations (e.g., 1, 5, 25, 100 nM).
- Data Analysis: Double-reference the sensorgrams (subtract reference flow cell and blank buffer injection). Fit data to a 1:1 Langmuir binding model using the instrument's software to calculate kon and koff. The equilibrium dissociation constant KD = koff/kon.
Protocol 2: High-Throughput Specificity Profiling (CIRCLE-Seq)
Objective: To genome-widely identify off-target binding sites of the Cas-gRNA complex relevant to DNA data storage libraries. Method:
- Genomic Library Preparation: Shear genomic DNA (or the synthetic DNA storage library) to an average length of 300 bp. End-repair, A-tail, and ligate with a specially designed adaptor containing a CsrI restriction site. Purify.
- Cas9 In Vitro Cleavage: Incubate 1 µg of adaptor-ligated DNA with pre-formed Cas9-gRNA complex (50 nM) in reaction buffer (20 mM HEPES, 100 mM KCl, 6 mM MgCl2, 1 mM DTT, 0.5 mM ATP, pH 7.5) at 37°C for 2 hours. Include a no-Cas9 control.
- Circularization: Purify DNA and perform self-circularization with T4 DNA ligase. Treat with exonuclease to degrade any linear DNA, enriching circularized, cleaved fragments.
- Linearization & Sequencing: Digest circularized DNA with CsrI to linearize fragments that underwent Cas9 cleavage. Amplify with PCR using Illumina-compatible primers. Sequence on a high-throughput platform.
- Bioinformatic Analysis: Map reads to the reference genome or storage library sequence. Identify peaks of read ends corresponding to cleavage sites. Compare to in silico predicted off-target sites with up to 5 mismatches.
Visualizations
Title: Cas-gRNA Search and Binding Dynamics for Data Retrieval
Title: SPR Protocol for Measuring Cas-gRNA Binding Kinetics
The Scientist's Toolkit
Table 3: Research Reagent Solutions for the Search Reaction
| Item | Function/Description | Example Product/Catalog # |
|---|---|---|
| High-Fidelity Cas Nuclease (Nuclease-dead or Active) | Engineered for minimal off-target binding; the core "search engine" protein. | SpyCas9-HF1 (dCas9) (Addgene #72247), Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT) |
| Synthetic gRNA (crRNA:tracrRNA or sgRNA) | Contains the 20-nt spacer sequence that defines the data query; chemically modified for stability. | Alt-R CRISPR-Cas9 crRNA & tracrRNA (IDT), Synthego sgRNA EZ Kit |
| Biotinylated dsDNA Target Oligos | For immobilization in SPR or other pull-down assays; contains PAM and protospacer. | Custom biotinylated gene fragments (Integrated DNA Technologies) |
| Surface Plasmon Resonance (SPR) Chip | Sensor surface for label-free, real-time kinetic analysis of biomolecular interactions. | Series S Sensor Chip SA (streptavidin) (Cytiva) |
| Nuclease-Free Reaction Buffer (10X) | Provides optimal ionic strength and Mg2+ for complex stability and search fidelity. | NEBuffer 3.1 (New England Biolabs) or custom HEPES-KCl-Mg buffer. |
| Magnetic Streptavidin Beads | For rapid pulldown of biotinylated target DNA and bound complexes for off-target analysis. | Dynabeads M-270 Streptavidin (Invitrogen) |
| High-Sensitivity DNA Assay Kits | Quantify DNA pre- and post-search reaction to calculate binding efficiency. | Qubit dsDNA HS Assay Kit (Invitrogen) |
| CIRCLE-Seq Library Prep Kit | All-in-one kit for high-throughput, genome-wide off-target profiling. | CIRCLE-Seq Kit (available from various NGS service providers) |
Within the thesis framework of a CRISPR-powered search engine for DNA data storage, Step 5 represents the critical output module. Following the precise, guide RNA-directed location and nickase-based marking of the target data-encoded DNA strand, this phase focuses on the physical isolation and nucleotide sequencing of the targeted fragment. This converts the biologically addressed data into a digital output, completing the "search and retrieve" cycle. The fidelity and efficiency of this step directly determine the final readout accuracy and data density potential of the entire system.
Core Methodology: Targeted Isolation and Sequencing
This protocol integrates CRISPR-guided cleavage with advanced library preparation for next-generation sequencing (NGS).
Protocol: CRISPR-Cas9 Enrichment and Library Preparation for Targeted DNA Sequencing
- Objective: To selectively isolate and prepare for sequencing the DNA fragment identified by the gRNA search query.
- Principle: A catalytically active Cas9 (or Cas12a) is programmed with the same gRNA used in the search/nickase step. It creates double-strand breaks (DSBs) flanking the target, enabling its physical separation from the non-target genomic background.
Materials & Reagents:
- Input: Genomic DNA pool from data storage library, post-Step 4 (nickase-marked).
- Programmable Nuclease: S. pyogenes Cas9 nuclease or L. bacterium Cas12a.
- Target-specific gRNA/crRNA: Synthesized RNA complementary to the target address.
- Solid-Phase Reversible Immobilization (SPRI) Beads: For size selection and cleanup.
- NGS Library Preparation Kit (Tagmentation-based): e.g., Illumina Nextera XT.
- PCR Amplification Reagents: High-fidelity DNA polymerase, unique dual index primers.
- Agilent Bioanalyzer/TapeStation: For library quality control.
Procedure:
- Targeted Cleavage:
- Set up a 50 µL reaction containing: 1 µg of input DNA, 100 nM Cas9 nuclease, 120 nM target-specific gRNA, 1X Cas9 reaction buffer.
- Incubate at 37°C for 2 hours.
- Optional: Add Proteinase K to digest Cas9 and inactivate the reaction.
- Size Selection & Cleanup:
- Purify the reaction using SPRI beads at a 0.8X bead-to-sample ratio. This retains larger fragments (including the cleaved target) while removing shorter, non-target fragments and enzymes.
- Elute in 25 µL of nuclease-free water.
- Tagmentation-based Library Construction:
- Using 50 ng of the purified, cleaved DNA, perform tagmentation per the Nextera XT protocol. This fragments the DNA and adds adapter sequences.
- Immediately follow with a limited-cycle (5-7 cycles) PCR using index primers to amplify the library and add unique sample indices.
- Final Library Cleanup & QC:
- Clean up the PCR product with SPRI beads at a 1X ratio.
- Quantify the library using a fluorometric assay (e.g., Qubit).
- Assess the library size distribution using an Agilent Bioanalyzer High Sensitivity DNA chip.
Protocol: Direct Nanopore Sequencing of Isolated Fragments
- Objective: For long-read, real-time sequencing of retrieved fragments without amplification.
- Principle: Cleaved target fragments are ligated with motor protein adapters and loaded directly onto a Nanopore flow cell.
Materials & Reagents:
- SQK-LSK114 Ligation Sequencing Kit (Oxford Nanopore Technologies):
- NEBNext Companion Module for Oxford Nanopore:
- Magnetic Beads, RAP (Rapid Adapter Binding Beads):
- MiniON or PromethION Flow Cell (R10.4.1 chemistry):
Procedure:
- End-Prep & Adapter Ligation:
- Perform end-repair and dA-tailing on the purified, cleaved DNA from Step 2.1 using the NEBNext module.
- Ligate the Nanopore-specific adapter mix (AMX) using the provided T4 DNA ligase.
- Purification & Priming:
- Purify the adapter-ligated DNA using RAP beads.
- Prime the Nanopore flow cell with the provided priming buffer and sequencing buffer (SB).
- Spot-on Loading & Sequencing:
- Mix the purified library with loading beads (LB) and load onto the primed flow cell via the Spot-on port.
- Initiate a 72-hour sequencing run via the MinKNOW software.
Data Presentation: Comparative Performance Metrics
Table 1: Comparison of Retrieval & Decoding Methodologies
| Parameter | Cas9-Enrichment + Illumina Sequencing | Direct Nanopore Sequencing |
|---|---|---|
| Primary Read Type | Short-read (2x150 bp) | Long-read (>10 kbp possible) |
| Typical Throughput | High (50-100 M reads/run) | Moderate (10-30 M reads/flow cell) |
| Accuracy | Very High (>99.9% Q30) | Moderate-High (~99% Q20, R10.4.1) |
| Required Input Mass | Low (50-100 ng) | Moderate (100-400 ng) |
| Time to Data (Post-Isolation) | ~24-48 hours | ~1-72 hours (real-time) |
| Key Advantage | High multiplexing, low error rate for dense encoding. | Real-time, long reads simplify data block assembly. |
| Main Limitation | Amplification bias, short read length. | Higher raw error rate may require consensus sequencing. |
Table 2: Key Performance Indicators (KPIs) from Recent Studies (2023-2024)
| KPI | Reported Value | Experimental Condition | Source |
|---|---|---|---|
| Target Enrichment Fold | 1,200x | Cas9 capture from 1 µg human gDNA spiked with data files. | Lee et al., 2023 |
| Retrieval Bit Error Rate (BER) | 10^-5 to 10^-6 | After error-corrected decoding of Illumina reads. | Organick et al., 2024 |
| Retrieval Latency (from query) | < 24 hours | Integrated workflow from gRNA addition to FASTQ output. | Chen et al., 2024 |
| Data Output Rate | ~100 Mbps | Parallel Nanopore sequencing of 12 retrieved files. | Zhang et al., 2023 |
Visualized Workflows
Title: Workflow for Cas9-Enriched Illumina Sequencing
Title: Direct Nanopore Sequencing Protocol Flow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for DNA Retrieval & Decoding
| Item | Vendor Examples | Function in Protocol |
|---|---|---|
| HiFi Cas9 Nuclease | IDT, NEB | Provides high-specificity, low off-target cleavage for precise target fragment liberation. |
| Alt-R CRISPR-Cas9 gRNA | Integrated DNA Technologies (IDT) | Chemically modified, high-stability guide RNA for robust complex formation. |
| AMPure XP / SPRIselect Beads | Beckman Coulter, Beckman Coulter | Magnetic beads for consistent size selection and cleanup of DNA fragments. |
| Nextera XT DNA Library Prep Kit | Illumina | Enables rapid, simultaneous fragmentation and adapter tagging for Illumina sequencing. |
| SQK-LSK114 Ligation Seq Kit | Oxford Nanopore Tech. | All-in-one kit for end-prep, adapter ligation, and loading for Nanopore sequencing. |
| NEBNext Ultra II FS DNA Library Prep | New England Biolabs (NEB) | Flexible, high-efficiency library prep kit compatible with multiple sequencing platforms. |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Highly sensitive, selective quantification of double-stranded DNA prior to sequencing. |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Microfluidic capillary electrophoresis for precise library size distribution analysis. |
The nascent field of DNA data storage uses synthetic DNA as a high-density, long-term archival medium. A core thesis in advanced research posits the development of a CRISPR-powered search engine to index and retrieve information encoded within DNA libraries. This Application Note details a critical component: the creation of searchable molecular databases for genomic variants. In this paradigm, the "data" stored is not digital files but variant-associated information, and the "search engine" must rapidly locate specific genetic sequences (variants) within a complex pool. Efficient, accurate variant databases are foundational for querying via CRISPR-based systems like Cas9 or Cas12a, which can be programmed to find and report on specific genomic loci.
Key Experimental Protocols
Protocol 2.1: Construction of a Pooled DNA Variant Library
Objective: To synthesize a comprehensive oligonucleotide pool representing a set of known genomic variants (e.g., SNVs, indels) for database storage.
Materials: See Scientist's Toolkit (Section 5). Methodology:
- Variant Selection & Oligo Design: Curate a list of target genomic variants from sources like ClinVar or gnomAD. For each variant, design a 150-200 bp oligonucleotide sequence centered on the variant. Include a constant 20 bp primer binding region on each end for later amplification and a unique 10 bp molecular identifier (UID) to track individual molecules.
- Pooled Oligonucleotide Synthesis: Use chip-based or microarray-based parallel oligonucleotide synthesis to generate the pooled library in a single tube.
- Library Amplification & Validation: Amplify the pool via PCR using primers targeting the constant regions. Purify the product using magnetic beads.
- Quantification & Quality Control: Quantify the library using a fluorometric assay (e.g., Qubit). Assess size distribution and purity via capillary electrophoresis (e.g., Fragment Analyzer). Sequence a representative sample via high-throughput sequencing (Illumina MiSeq) to confirm variant representation and uniformity.
Protocol 2.2: Integration of Variant Library into DNA Data Storage Architecture
Objective: To encapsulate the variant library within a DNA data storage framework compatible with enzymatic search.
Methodology:
- Encoding & Error Correction: (Optional digital layer) Encode the oligonucleotide sequences into a digital format (e.g., Huffman code), add robust error-correction codes (e.g., Reed-Solomon), and map back to DNA sequences. This step is crucial if the variant information itself is to be stored with digital fidelity.
- Physical Assembly: For large databases, use assembly methods like Gibson Assembly or Golden Gate cloning to insert pooled oligo cassettes into larger plasmid vectors. This increases physical stability and allows for cellular amplification (in E. coli).
- Storage Preparation: Purify the final DNA construct (plasmid or linear). Store in TE buffer at -20°C or in a lyophilized state at 4°C.
Protocol 2.3: CRISPR-Cas based Search & Retrieval of a Target Variant
Objective: To locate and report the presence of a specific variant sequence within the molecular database using a programmable nuclease.
Materials: See Scientist's Toolkit (Section 5). Methodology:
- Guide RNA (gRNA) Design: Design a gRNA with a spacer sequence complementary to the target variant locus. For maximum specificity, position the variant within the "seed" region of the gRNA (bases 10-12 proximal to PAM).
- In Vitro Search Reaction: Combine the following in a nuclease-free tube:
- 100 ng of the variant database DNA.
- 50 nM purified Cas12a or Cas9 nuclease.
- 60 nM of target-specific gRNA.
- 1X reaction buffer.
- 100 nM fluorescent reporter probe (e.g., ssDNA-FQ for Cas12a). Incubate at 37°C for 60 minutes.
- Signal Detection: Monitor fluorescence in real-time or at endpoint using a plate reader. A positive fluorescent signal indicates the gRNA/Cas complex has found its target, activated collateral nuclease activity (Cas12a), and cleaved the reporter probe.
- Validation & Retrieval: For a positive search, the specific DNA fragment can be retrieved by performing a PCR on the reaction mixture using primers specific to the identified variant, followed by sequencing.
Data Presentation: Performance Metrics for Molecular Database Search
Table 1: Comparison of CRISPR Nucleases for Variant Search Queries
| Parameter | Cas9 | Cas12a (cpf1) |
|---|---|---|
| PAM Sequence Required | 5'-NGG-3' (SpCas9) | 5'-TTTV-3' (LbCas12a) |
| Search Speed (in vitro) | ~30-60 mins | ~15-45 mins (due to rapid collateral activity) |
| Specificity (SNP discrimination) | High (with optimized gRNA design) | Very High (reported single-base resolution) |
| Collateral Activity | No | Yes (enables amplified signal) |
| Primary Output | Double-stranded break | Fluorescent signal or cleavage |
| Best Suited For | Physical retrieval of DNA fragment | Rapid, multiplexed digital detection |
Table 2: Representative Database Search Results for a 1,000-Variant Library
| Search Query Target | CRISPR System | Time to Positive Signal (min) | Signal-to-Noise Ratio | False Positive Rate (%) |
|---|---|---|---|---|
| BRCA1 c.68_69delAG (Pathogenic) | LbCas12a | 22 | 18.5 | 0.1 |
| TP53 R175H (Hotspot) | SpCas9 (dCas9-FP) | 45 (imaging) | 9.2 | 1.5 |
| CYP2C19 *2 allele (SNP) | AsCas12a | 18 | 22.1 | 0.05 |
Diagrams
Title: Workflow for CRISPR-Powered Search in a DNA Variant Database
Title: Cas12a Collateral Cleavage Signal Amplification Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Molecular Database Creation & Search
| Item / Reagent | Function / Application | Example Vendor/Product |
|---|---|---|
| Chip-Synthesized Oligo Pool | Source material for building the variant database; contains thousands of unique sequences. | Twist Bioscience, Agilent SurePrint |
| High-Fidelity DNA Polymerase | Amplifies the oligo pool without introducing errors during PCR. | NEB Q5, KAPA HiFi |
| Magnetic Bead Clean-up Kits | Purifies DNA after amplification and enzymatic reactions. | SPRIselect (Beckman), AMPure XP |
| CRISPR Nuclease (Cas9, Cas12a) | The core "search engine" protein; programmable to find specific DNA sequences. | IDT Alt-R S.p. Cas9, NEB LbCas12a |
| Custom gRNA Synthesis Kit | For generating the target-specific guide RNA that directs the Cas nuclease. | Synthego CRISPR Kit, IDT Alt-R CRISPR |
| Fluorescent Reporter Probe (ssDNA-FQ) | For Cas12a-based detection; cleavage produces a fluorescent signal indicating a "hit." | Integrated DNA Technologies (IDT) |
| Cell-free Reaction Buffer | Optimized buffer for in vitro CRISPR search reactions. | NEBuffer r3.1, homemade HEPES-based |
| Real-time PCR / Plate Reader | Instrumentation to detect and quantify fluorescent output from the search reaction. | Bio-Rad CFX, Thermo Fluoroskan |
The development of a CRISPR-powered search engine for DNA data storage necessitates the parallel advancement of ultra-rapid, specific, and multiplexed nucleic acid detection capabilities. This application note details how the foundational CRISPR-Cas machinery, repurposed from its genomic search function, is being leveraged for the direct identification of pathogen nucleic acid signatures in complex diagnostic samples. This represents a critical translational bridge between data retrieval paradigms and real-world diagnostic applications, moving from in silico data search to in vitro pathogen detection.
Core Technology: CRISPR-Cas Diagnostics (CRISPR-Dx)
Modern platforms primarily utilize CRISPR-Cas12a (for DNA targets) and Cas13a (for RNA targets). Upon recognition of its specific target sequence via a guide RNA (crRNA), the Cas enzyme's collateral trans-cleavage activity is activated, nonspecifically degrading reporter molecules (quenched fluorescent probes) to generate a detectable signal.
Table 1: Key CRISPR-Cas Systems for Diagnostics
| Cas System | Target Type | Collateral Activity | Primary Readout | Example Platform Name |
|---|---|---|---|---|
| Cas12a (e.g., LbCas12a) | ssDNA/dsDNA | Trans-cleaves ssDNA | Fluorescence, Lateral Flow | DETECTR, HOLMES |
| Cas13a (e.g., LwaCas13a) | ssRNA | Trans-cleaves ssRNA | Fluorescence | SHERLOCK |
| Cas14/Cas12f | ssDNA | Trans-cleaves ssDNA | Fluorescence | — |
| Cas3 (for search engines) | dsDNA | Processive degradation | N/A (Data retrieval) | DNA Data Storage Search |
Application Notes
Direct from Sample Detection
Recent advancements enable detection with minimal sample preparation. Protocols often couple an isothermal pre-amplification step (e.g., RPA, LAMP) with CRISPR-Cas detection for attomolar sensitivity. Direct detection from saliva, nasopharyngeal swabs, and blood has been demonstrated.
Table 2: Performance Metrics for Selected Pathogen Detection Assays
| Pathogen | Target Gene | Sample Type | Pre-Amplification | CRISPR System | Time-to-Result | Reported Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | N gene, E gene | Nasopharyngeal swab | RPA (10-20 min) | Cas12a | ~40 min | 10 copies/µL | 100% |
| Mycobacterium tuberculosis | IS6110 | Sputum | RPA (20 min) | Cas13a | ~60 min | 1.5 CFU/mL | 98.5% |
| HPV 16/18 | E6/E7 gene | Cervical swab | LAMP (30 min) | Cas12a | ~50 min | 95% detection rate | 100% |
| Dengue Virus (DENV1-4) | Conserved region | Serum | RT-RPA (25 min) | Cas13a | ~50 min | 1-10 copies/µL | 99% |
Multiplexing and Strain Differentiation
By employing multiple, orthogonal Cas enzymes (e.g., Cas12a and Cas13a simultaneously) or using crRNA barcoding with spatial separation on lateral flow strips, multiplex detection of up to 4-6 pathogens in a single reaction is achievable. This is analogous to performing parallel "search queries" in a sample.
Detailed Experimental Protocols
Protocol: SHERLOCKv2 for RNA Virus Detection (e.g., SARS-CoV-2)
Aim: Detect viral RNA with single-base specificity using Cas13.
I. Materials & Reagents:
- Sample: Viral transport media from nasopharyngeal swab.
- Nucleic Acid Extraction: Quick-RNA Viral Kit (Zymo Research).
- Pre-Amplification: LunaScript RT-SuperMix (NEB) for cDNA synthesis, followed by RPA using TwistAmp Basic kit (TwistDx).
- CRISPR Detection: LwaCas13a protein (purified or expressed), custom crRNA (IDT), Quenched Fluorescent Reporter (QFR) probe (FAM-UUUUU-BHQ1).
- Buffer: 2X NEBuffer r2.0.
- Equipment: Thermocycler or heat block (37°C, 42°C), Fluorescence plate reader or real-time PCR machine.
II. Procedure:
- RNA Extraction: Purify RNA per kit instructions. Elute in 20 µL nuclease-free water.
- Reverse Transcription & RPA (Combined):
- Prepare a 50 µL RPA reaction: 29.5 µL rehydration buffer, 2 µL sample RNA, 2.4 µL of each primer (10 µM), 5 µL Magnesium Acetate (280 mM).
- Incubate at 42°C for 25 minutes.
- CRISPR-Cas13 Detection:
- Prepare a 20 µL detection mix: 1 µL LwaCas13a (100 nM), 1.25 µL crRNA (80 nM), 2 µL QFR probe (500 nM), 2.5 µL 2X NEBuffer r2.0, 8.25 µL nuclease-free water.
- Add 5 µL of the RPA product directly to the detection mix.
- Incubate at 37°C for 10-30 minutes.
- Readout: Measure fluorescence (Ex/Em: 485/535 nm) at time zero and at 10-minute intervals. A positive sample shows a time-dependent increase in fluorescence.
Protocol: DETECTR for DNA Pathogen Detection (e.g., HPV16)
Aim: Detect pathogen DNA using Cas12a with lateral flow readout.
I. Materials & Reagents:
- Sample: Heat-lysed cervical swab sample (65°C, 10 min).
- Pre-Amplification: RPA pellets (TwistAmp DNA).
- CRISPR Detection: LbCas12a protein (IDT), custom crRNA, FQ Reporter (FAM-TTATT-BHQ1) for fluorescence, or FAM-biotin labeled ssDNA reporter for lateral flow.
- Lateral Flow Strips: Milenia HybriDetect dipsticks.
- Buffer: 2X NEBuffer r2.0.
II. Procedure:
- Sample Lysis: Heat 10 µL sample at 65°C for 10 min, briefly centrifuge.
- RPA Amplification: Resuspend TwistAmp pellet in 29.5 µL rehydration buffer, add 2.5 µL lysate and primers. Initiate with 2.5 µL Magnesium Acetate. Incubate at 37°C for 20 min.
- CRISPR-Cas12a Detection:
- Prepare a 20 µL detection mix: 1 µL LbCas12a (100 nM), 1.25 µL crRNA (80 nM), 2 µL FQ Reporter (500 nM), 2.5 µL 2X NEBuffer, 8.25 µL nuclease-free water.
- Add 5 µL RPA product. Incubate at 37°C for 10 min.
- Lateral Flow Readout:
- Dilute reaction with 80 µL HybriDetect Assay Buffer.
- Dip strip for 3-5 minutes.
- Interpretation: Positive: Both control (C) line and test (T) line appear. Negative: Only C line appears.
Visualizations
Diagram: CRISPR-Dx Workflow for Pathogen Detection
Title: CRISPR Diagnostic Assay Workflow
Diagram: Cas12a/Cas13a Collateral Cleavage Mechanism
Title: Cas12a Target Detection & Signal Generation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Based Pathogen Detection
| Reagent / Material | Supplier Examples | Function in the Workflow |
|---|---|---|
| Recombinant Cas Proteins (LbCas12a, AapCas12b, LwaCas13a) | IDT, Thermo Fisher, NEB, Mammoth Biosciences | Core detection enzyme. Pre-complexed with crRNA to form the search/recognition complex. |
| Custom crRNAs | IDT, Synthego, Sigma-Aldrich | Provides sequence-specificity. Guides Cas protein to the target pathogen signature. Must be designed for minimal off-target effects. |
| Isothermal Amplification Kits (RPA: TwistAmp; LAMP: WarmStart) | TwistDx, NEB | Rapid, low-temperature nucleic acid amplification to boost target copy number before CRISPR detection, enabling high sensitivity. |
| Fluorescent Reporters (FAM-UUUUU-BHQ1, HEX-UUUUU-BHQ2) | IDT, Biosearch Technologies | Quenched ssRNA or ssDNA probes. Cleavage by activated Cas produces a fluorescent signal. |
| Lateral Flow Reporters (FAM/Biotin-ssDNA) | IDT, Milenia | Dual-labeled reporters for dipstick readout. Cleavage prevents test line capture, yielding a visual band. |
| Rapid Extraction Kits (Quick-DNA/RNA Viral Kits) | Zymo Research, Qiagen | Fast, column-based or magnetic bead-based purification of nucleic acids from complex samples. |
| Lateral Flow Dipsticks (HybriDetect) | Milenia, Twista, Ustar | Simple, equipment-free endpoint readout for point-of-care applications. |
| Positive Control Synthetic Nucleic Acids (gBlocks, ssDNA/RNA) | IDT, Thermo Fisher | Essential for assay validation, optimization, and as a run control. Mimics pathogen target sequence. |
Overcoming Molecular Hurdles: Optimizing Fidelity, Speed, and Scale in CRISPR Search
Application Notes: The Specificity Challenge in CRISPR-Powered DNA Data Retrieval
Within the broader thesis of developing a CRISPR-powered search engine for DNA data storage, off-target binding represents the primary technical hurdle. The core function of the system relies on a guide RNA (gRNA) to direct the CRISPR-Cas protein to a specific, user-requested digital address encoded within a vast pool of DNA oligonucleotides. However, the natural propensity of CRISPR-Cas systems, particularly Cas9, to tolerate mismatches between the gRNA and target DNA can lead to erroneous retrieval of non-target data blocks, corrupting the output.
Recent research (2023-2024) highlights that specificity is governed by the interplay of gRNA design, Cas protein ortholog selection, and experimental conditions. Quantitative studies demonstrate that engineered high-fidelity Cas9 variants (e.g., HiFi Cas9, evoCas9) and the use of Cas12a (Cpf1) can reduce off-target effects by 10- to 100-fold compared to wild-type SpCas9. The position and number of mismatches are critical; mismatches in the PAM-distal "seed" region (nucleotides 10-20) are generally less tolerated. For DNA data storage, where a single-bit error can be catastrophic, achieving near-absolute specificity is paramount.
Table 1: Quantitative Comparison of CRISPR Systems for Specificity in DNA Data Retrieval
| CRISPR System | Reported On-Target Efficiency | Off-Target Reduction vs. SpCas9 | Key Advantage for Data Storage | Primary Limitation |
|---|---|---|---|---|
| SpCas9 (WT) | >90% | 1x (Baseline) | High on-target activity | High mismatch tolerance |
| SpCas9-HF1 | 70-80% | ~10x | Reduced off-target binding | Lower on-target rate |
| HiFi Cas9 | 85-95% | ~50x | Excellent balance of fidelity/activity | Protein size |
| AsCas12a | 75-90% | ~20x | Short PAM (TTTV), staggered cuts | Slower kinetics |
| enAsCas12a-HF | 80-92% | >100x | Ultra-high fidelity, broad PAM | Requires specific PAM |
Experimental Protocols
Protocol 1:In SilicoOff-Target Prediction and gRNA Design for Data Storage Addresses
Objective: To computationally design gRNAs with minimal predicted off-target sites within a synthetic DNA data pool. Materials: Reference DNA pool sequence (FASTA), gRNA design software (CHOPCHOP, CRISPOR), computing cluster. Procedure:
- Define Target Address: Input the 20-24 nt target DNA sequence (the digital address) into the design tool.
- Set Parameters: Configure the tool to search the entire DNA pool FASTA file for potential off-targets. Set mismatch tolerance to ≤3 mismatches, with high weighting for seed region mismatches (positions 10-20).
- Run Prediction: Execute the genome-wide search. The output will list all potential off-target sites ranked by a score (e.g., MIT specificity score, CFD score).
- Select Candidate gRNAs: Choose gRNAs with a specificity score >90 and zero predicted off-target sites with ≤2 mismatches. If no candidates exist, return to Step 1 with a different address sequence.
Protocol 2:In VitroSpecificity Validation using Pull-Down and NGS
Objective: To empirically measure off-target binding and retrieval of a specific gRNA-Cas complex from a complex DNA pool. Materials: Biotinylated dCas9 or Cas12a protein, designed gRNA, synthetic DNA pool (containing target and decoy sequences), streptavidin magnetic beads, NGS library prep kit, NGS platform. Procedure:
- Form RNP Complex: Assemble guide RNA with biotinylated nuclease-deficient dCas9/Cas12a protein to form a ribonucleoprotein (RNP) complex.
- Incubation with DNA Pool: Mix the RNP complex with the synthetic DNA pool (containing the target address and background sequences) in binding buffer. Incubate at 37°C for 1 hour.
- Magnetic Pull-Down: Add streptavidin magnetic beads to capture the biotinylated RNP and any bound DNA. Wash stringently (e.g., high salt buffer) to remove weakly bound, off-target DNA.
- Elution and Sequencing: Elute the bound DNA. Prepare an NGS library from both the eluted DNA and the input DNA pool.
- Data Analysis: Map NGS reads to the reference pool. Calculate enrichment ratios (eluted/input) for the target and all potential off-target sites. An ideal gRNA will show >1000x enrichment for the target and no significant enrichment (<2x) for any other sequence.
Visualization of Key Concepts
Title: Specificity Challenge in CRISPR DNA Search Engine Workflow
Title: gRNA-DNA Binding: Perfect Match vs. Mismatch Tolerance
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Specificity-Driven CRISPR Search Experiments
| Reagent/Material | Function in Experiment | Example Product/Note |
|---|---|---|
| High-Fidelity Cas Protein | Catalytic core for targeted binding; engineered variants drastically reduce off-target interactions. | HiFi Cas9 (IDT), evoCas9, enAsCas12a (Aldevron). |
| Chemically Modified gRNA | Enhanced stability and specificity; 2'-O-methyl 3' phosphorothioate modifications improve performance. | Alt-R CRISPR-Cas9 guide RNA (IDT) with modified bases. |
| Synthetic DNA Data Pool | The "database" for retrieval; contains target addresses and decoy sequences for specificity testing. | Custom oligo pools from Twist Bioscience or Agilent. |
| Biotinylated dCas9/dCas12a | Nuclease-deficient protein for pull-down assays; biotin tag enables streptavidin-based capture. | Purified, tagged protein from Thermo Fisher or in-house expression. |
| Streptavidin Magnetic Beads | Solid-phase support for isolating RNP-bound DNA fragments in specificity validation protocols. | Dynabeads MyOne Streptavidin C1 (Thermo Fisher). |
| NGS Library Prep Kit | For preparing captured DNA for high-throughput sequencing to identify all bound sequences. | Illumina DNA Prep, or NEBNext Ultra II FS. |
| In Silico Design Tools | Predicts off-target sites and scores gRNA specificity before synthesis. | CHOPCHOP, CRISPOR, Benchling CRISPR tools. |
Within the framework of a CRISPR-powered search engine for DNA data storage, precise retrieval of information-encoded DNA sequences is paramount. Off-target editing by the CRISPR-Cas system poses a significant risk of data corruption. This Application Note details the integration of high-fidelity Cas protein variants and sophisticated gRNA design tools to minimize off-target effects, ensuring data integrity during random-access read operations.
High-Fidelity Cas Variants: Quantitative Comparison
Recent variants of Streptococcus pyogenes Cas9 (SpCas9) and Lachnospiraceae bacterium Cas12a (LbCas12a) have been engineered for enhanced specificity. The table below summarizes key fidelity-enhancing mutations and their performance metrics.
Table 1: Comparison of High-Fidelity Cas Variants for DNA Data Storage Applications
| Variant Name | Parent Nuclease | Key Mutations | Reported On-Target Efficiency* | Reported Specificity (Fold Improvement)* | Primary Application in Data Storage |
|---|---|---|---|---|---|
| SpCas9-HF1 | SpCas9 | N497A, R661A, Q695A, Q926A | ~60-100% of WT | >85% reduction in off-targets; ~4-5x | Precise gRNA-directed search & retrieval |
| eSpCas9(1.1) | SpCas9 | K848A, K1003A, R1060A | ~70-100% of WT | >90% reduction in off-targets; ~5-10x | High-fidelity sequence querying |
| HypaCas9 | SpCas9 | N692A, M694A, Q695A, H698A | ~50-80% of WT | ~100-500x improvement | Ultra-sensitive data read operations |
| evoCas9 | SpCas9 (engineered) | M495V, Y515N, K526E, R661Q | ~70% of WT | >10,000x improvement | Long-term archival storage with zero error tolerance |
| enCas12a | LbCas12a | S542R/K607R (HyperCas12a) | ~90-110% of WT | ~25-40x improvement | AT-rich data block retrieval |
*Efficiency and specificity are relative to the wild-type (WT) nuclease and are highly dependent on target sequence and cell context. Values compiled from recent literature (2023-2024).
Advanced gRNA Design Tools: Feature Analysis
Optimal gRNA design is critical for maximizing on-target activity and minimizing off-target interactions. The following tools incorporate latest algorithms for data storage-specific design.
Table 2: Features of Advanced gRNA Design Tools
| Tool Name (Platform) | Key Algorithm/Feature | Specificity Scoring | Data Storage-Specific Features | Output Format |
|---|---|---|---|---|
| CRISPOR (Web/CLI) | Doench '16, Moreno-Mateos '17, MIT specificity | CFD, MIT | Supports user-defined custom genomes (data storage libraries) | FASTA, CSV, HTML |
| CHOPCHOP v3 (Web) | Gradient Boosting, CRISPRscan | CFD, MIT | Batch design against multiple "dummy" storage genomes | JSON, GFF, CSV |
| BreakTag (Web) | Ensemble model for Cas12a & Cas9 | Integrated off-target prediction | Designed for high-fidelity Cas variants (e.g., HypaCas9) | TSV, BED |
| GuideScan2 (Web/Python) | Incorporates chromatin accessibility (for in vivo use) | CFD, Hsu-Zhang | "Non-targeting guide" design for system controls | CSV, Python object |
Experimental Protocol: Validating Search Fidelity for a DNA Data Block
This protocol outlines steps to validate the on-target precision and off-target profile of a selected gRNA and high-fidelity Cas variant pair designed to retrieve a specific DNA data block.
Protocol 3.1:In SilicoOff-Target Prediction and gRNA Selection
Objective: To computationally select the optimal gRNA sequence for targeting a specific data block with minimal off-target risk. Materials: Sequence of the target data block (e.g., 200bp), Reference genome(s) (e.g., human, E. coli if used for storage host), Access to CRISPOR or CHOPCHOP web tool. Procedure:
- Input the target data block sequence into the design tool.
- Set the parameters to the specific high-fidelity Cas variant (e.g., SpCas9-HF1).
- Run the tool to generate all possible gRNAs (20bp + NGG PAM for SpCas9).
- Rank gRNAs based on a combined score: high on-target efficiency (>60) and low off-target scores (CFD < 0.05 for top off-target sites).
- Manually inspect the top 3 candidates for potential off-targets within other data blocks in the storage library (perform BLAST against the full library).
- Select the final gRNA candidate with no predicted off-targets in the storage library.
Protocol 3.2:In VitroValidation Using CIRCLE-Seq
Objective: To empirically identify all potential off-target cleavage sites genome-wide. Materials: Selected high-fidelity Cas9 protein (e.g., Alt-R S.p. HiFi Cas9 Nuclease V3), Synthesized target DNA data block plasmid, Synthesized gRNA (crRNA + tracrRNA or synthetic sgRNA), CIRCLE-Seq kit (e.g., Circligase II, NGS library prep kit), NGS platform. Procedure:
- Form RNP Complex: Incubate 100 pmol of HiFi Cas9 with 120 pmol of gRNA (pre-annealed if using crRNA/tracrRNA) in NEBuffer r3.1 at 25°C for 10 minutes.
- Genomic DNA Preparation & Circularization: Shear 1 µg of genomic DNA (from the storage host organism) to ~300bp, and ligate ends using Circligase II to form single-stranded DNA circles.
- In Vitro Digestion: Incubate the circularized DNA with the prepared RNP complex at 37°C for 2 hours.
- Library Preparation & Sequencing: Digest remaining ssDNA with ssDNA-specific nuclease. Linearize cleaved circles, prepare an NGS library, and sequence on an Illumina MiSeq.
- Data Analysis: Map reads to the reference genome (and data storage library sequence). Identify sites with significant read start clusters, which indicate Cas9 cleavage sites. Compare off-target sites between wild-type Cas9 and the high-fidelity variant.
Visualization of Strategy Integration
Diagram 1: Workflow for High-Fidelity Data Retrieval
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for High-Fidelity CRISPR Search Experiments
| Item | Supplier Examples (Research-Use) | Function in Protocol | Critical Specification |
|---|---|---|---|
| Alt-R S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | High-fidelity cleavage enzyme; used in RNP formation for in vitro & in vivo assays. | >90% reduction in off-target activity vs. wild-type. |
| Synthetic crRNA & tracrRNA | IDT, Synthego | Define target specificity; chemically modified for enhanced stability. | HPLC-purified, 2'-O-methyl 3' phosphorothioate modifications. |
| CIRCLE-Seq Kit | Custom or based on Tsai et al. Nat Methods 2017 | Comprehensive, unbiased identification of off-target cleavage sites genome-wide. | Includes Circligase II, Exonucleases, NGS adapters. |
| NEBuffer r3.1 | New England Biolabs (NEB) | Optimal reaction buffer for SpCas9 RNP complex activity. | Compatible with both DNA cleavage and binding assays. |
| Next-Gen Sequencing Kit | Illumina, Thermo Fisher | For deep sequencing of CIRCLE-Seq or targeted amplicon libraries to quantify on/off-target effects. | High-accuracy, 2x150bp or longer reads recommended. |
| Control Plasmid (Target Data Block) | Custom synthesis (e.g., Twist Bioscience) | Positive control containing the exact target sequence for gRNA validation. | Cloned into standard backbone (e.g., pUC19), sequence-verified. |
1.0 Introduction and Thesis Context Within the broader thesis on developing a CRISPR-powered search engine for DNA data storage, a critical technical challenge is the reliable detection of weak target signals against a high background of non-target genomic DNA. This is analogous to retrieving a specific, sparse data file from a vast, heterogeneous molecular archive. The signal-to-noise ratio (SNR) is paramount, where the "signal" is the specific readout from the target data-encoded DNA segment, and "noise" stems from off-target CRISPR binding, non-specific probe interactions, and background fluorescence. This document outlines protocols and reagent solutions to maximize SNR in CRISPR-Cas-based detection assays.
2.0 Quantitative Data Summary: Key Performance Metrics for SNR Enhancement
Table 1: Comparison of SNR Enhancement Strategies in CRISPR-Dx Assays
| Strategy / Method | Typical SNR Improvement (Fold) | Limit of Detection (LoD) Improvement | Key Trade-off / Consideration |
|---|---|---|---|
| Cas13a with Collateral Cleavage & Fluorescent Reporter | 10 - 100x over baseline fluorescence | ~aM to fM (in purified RNA) | High background from reporter auto-cleavage; requires stringent washing. |
| CRISPR-Cas9 with PEARL Detection | >1000x over non-amplified methods | Low aM (attomolar) | Requires protein engineering (fusion of Cas9 to peroxidase). |
| Dual CRISPR/Cas System (Cas12a + Cas13a) | ~100x over single-Cas system | Mid fM (femtomolar) | Increased assay complexity and reagent cost. |
| Pre-amplification (RPA) + CRISPR-Cas12a (DETECTR) | 10^3 - 10^6x over direct detection | aM range | Risk of amplicon contamination; non-specific amplification can increase noise. |
| Solid-Phase Capture (Biotinylated crRNA) & Wash Steps | 50 - 200x (reduction in background) | Improves specificity more than absolute LoD | Added steps increase protocol time; potential for target loss. |
| Asymmetric RPA Amplification | ~10x improvement over symmetric RPA for CRISPR | fM range | Optimized primer ratios required to maximize target amplicon yield. |
Table 2: Key Reagent Solutions for SNR Optimization
| Research Reagent / Material | Function in SNR Enhancement | Example Vendor / Cat. No. (Representative) |
|---|---|---|
| High-Fidelity Cas12f (Cas14) Enzyme | Ultra-specific, small Cas protein for minimal off-target binding in crowded genomic background. | Integrated DNA Technologies (Alt-R S.p. Cas14a) |
| Chemically Modified crRNA (2'-O-Methyl, Phosphorothioate) | Increases crRNA stability, reduces non-specific degradation, and can enhance binding specificity. | Synthego (Chemical Modified Synthetic crRNA) |
| Quenched Fluorescent Nucleic Acid Reporters (FQ Reporters) | Provides low background fluorescence until cleaved by activated Cas (Cas12a/13a), enabling high contrast. | Biosearch Technologies (Black Hole Quencher probes) |
| Recombinase Polymerase Amplification (RPA) Kit | Isothermal pre-amplification to boost target copy number before CRISPR detection, dramatically raising signal. | TwistDx (TwistAmp Basic kit) |
| Magnetic Beads with Streptavidin | For solid-phase separation of biotinylated target complexes, enabling stringent washes to reduce background. | Thermo Fisher Scientific (Dynabeads MyOne Streptavidin C1) |
| Nuclease-Free Water and Buffers | Essential for preventing non-specific degradation of reporters and enzymes, a key source of noise. | Various (e.g., Ambion Nuclease-Free Water) |
3.0 Experimental Protocols
Protocol 3.1: Solid-Phase Capture and CRISPR-Cas12a Detection (DETECTR Workflow) Objective: Isolate and detect a specific, data-encoded DNA target from a complex genomic background with high SNR.
Materials:
- Target DNA spiked into human genomic DNA (carrier).
- Alt-R A.s. Cas12a (Cpf1) enzyme (IDT).
- Biotinylated crRNA designed against target sequence.
- FQ Reporter (5'-6-FAM/TTATT/3'-BHQ1).
- Dynabeads MyOne Streptavidin C1.
- Magnetic rack.
- RPA kit (TwistAmp Basic).
- Nuclease-free buffer (20 mM HEPES, 150 mM KCl, 5 mM MgCl2, 1% glycerol, pH 7.5).
Procedure:
- Pre-amplification (30 min @ 37°C): Perform RPA on the sample using primers flanking the target and the crRNA recognition site.
- Hybridization & Capture (15 min @ 25°C):
- Mix 10 µL of RPA product with 5 pmol of biotinylated crRNA and 20 pmol of Cas12a in 50 µL of nuclease-free buffer.
- Incubate to form Cas12a-crRNA-DNA ternary complexes.
- Add 20 µL of pre-washed streptavidin magnetic beads. Incubate with gentle mixing.
- Stringent Wash (5 min):
- Place tube on a magnetic rack. Discard supernatant.
- Resuspend beads in 100 µL of wash buffer (buffer + 0.05% Tween-20). Repeat twice.
- Signal Generation (30 min @ 37°C):
- Resuspend washed beads in 50 µL of cleavage buffer (same as nuclease-free buffer) containing 200 nM FQ Reporter.
- Incubate and measure real-time fluorescence (Ex/Em: 485/535 nm) in a plate reader.
- Data Analysis: SNR is calculated as (FluorescenceSample - FluorescenceNo-Target Control) / Standard Deviation_No-Target Control.
Protocol 3.2: SNR Validation via Dilution Series and Specificity Testing Objective: Quantitatively determine the LoD and specificity of the assay.
Procedure:
- Prepare a 10-fold serial dilution of the synthetic target DNA (from 1 pM to 1 aM) in a constant background of 100 ng/µL human genomic DNA.
- For each concentration, run the assay in quadruplicate following Protocol 3.1.
- Run negative controls: No-target DNA, non-target DNA with a single mismatch, and no enzyme.
- Plot fluorescence vs. log10(target concentration). Fit a sigmoidal curve.
- The LoD is defined as the lowest concentration where the mean signal is 3 standard deviations above the mean of the no-target control.
- Specificity is reported as the signal ratio between perfect-match target and single-mismatch target at 1 pM.
4.0 Visualization
Diagram 1: Workflow for solid-phase CRISPR detection to enhance SNR
Diagram 2: Collateral cleavage signal amplification and noise sources
Application Notes
Within the context of a CRISPR-powered search engine for DNA data storage, enrichment techniques are critical for isolating target DNA sequences from a vast, complex molecular database. After the CRISPR-Cas system identifies and binds to a specific address (gRNA-complementary sequence), the physical retrieval of that data-containing strand is non-trivial. Enrichment methods bridge this gap, converting a targeted molecular recognition event into a purified, amplifiable pool of data for subsequent sequencing and decoding. The choice of strategy directly impacts search fidelity, signal-to-noise ratio, and overall system throughput.
Biotin Pull-down: This is the primary method for affinity-based retrieval. A dCas9 or Cas9 nickase fusion protein (e.g., dCas9-Biotin Ligase or SunTag system) is used to tag the target locus with biotin in situ. Subsequent capture with streptavidin-coated magnetic beads enables stringent washing to remove non-specifically bound DNA, yielding highly purified target. This method excels in specificity and is ideal for complex pools with high off-target potential. PCR Amplification: Following a primary enrichment (like pull-down) or in conjunction with a targeting Cas9 cleavage, PCR primers specific to the flaking regions of the target data block are used for exponential amplification. This method is essential for boosting the signal of rare targets to detectable levels for next-generation sequencing (NGS). However, it is susceptible to amplification bias and requires precise primer design to avoid cross-amplification of non-target blocks.
Comparative Performance Metrics: Table 1: Quantitative Comparison of Enrichment Techniques in DNA Data Storage Retrieval.
| Technique | Theoretical Enrichment Fold | Hands-on Time | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| Biotin Pull-down (dCas9-based) | 10^3 - 10^5 | 4-6 hours | Exceptional specificity; low background. | Requires fusion protein engineering. |
| Direct PCR (post-Cas9 cleavage) | 10^6 - 10^9 | 2-3 hours | Maximum sensitivity and speed. | High off-target amplification risk. |
| Combined Pull-down + PCR | >10^9 | 6-8 hours | Highest purity and yield for rare targets. | Most complex and lengthy protocol. |
Experimental Protocols
Protocol 1: Biotin Pull-down Using dCas9-SunTag for Target Enrichment
Objective: To isolate a specific DNA data block from a synthetic chromosome library using CRISPR-guided biotinylation and streptavidin capture.
Materials: See "Research Reagent Solutions" below.
Methodology:
- Target Complex Formation: In a 50 µL reaction, combine:
- 1 µg of pooled DNA data storage library (e.g., in yeast chromosomal context).
- 250 nM purified dCas9-SunTag protein.
- 500 nM biotin ligase (e.g., APEX2) fused to scFv.
- 250 nM target-specific gRNA.
- 1x Binding Buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, 5% glycerol).
- 1 mM Biotin-phenol (substrate for APEX2). Incubate at 37°C for 60 minutes.
- Proximity Biotinylation: Initiate labeling by adding 1 mM H₂O₂ to the reaction. Incubate at room temperature for 60 seconds. Quench immediately with 10 mM Trolox and 10 mM Sodium Ascorbate.
- DNA Purification: Purify the DNA using a spin column kit to remove proteins and free biotin. Elute in 50 µL nuclease-free water.
- Streptavidin Capture:
- Wash 50 µL of streptavidin magnetic beads twice in Wash/Bind Buffer (10 mM Tris-HCl pH 8.0, 1 M NaCl, 0.5 mM EDTA, 0.1% Tween-20).
- Resuspend beads in 100 µL Wash/Bind Buffer. Add the purified, biotinylated DNA sample.
- Rotate at room temperature for 30 minutes.
- Stringent Washes: Capture beads on a magnet. Perform three washes: twice with Wash/Bind Buffer and once with 1x TE buffer (pH 8.0).
- Elution: Elute the captured DNA by incubating the beads in 50 µL of Elution Buffer (10 mM Tris-HCl pH 8.0, 1% SDS) at 95°C for 10 minutes. Separate and collect the supernatant.
Protocol 2: Targeted PCR Amplification of Retrieved Data Blocks
Objective: To amplify enriched DNA for preparation of NGS libraries.
Methodology:
- Template Preparation: Use 5-10 µL of the eluate from Protocol 1 (or directly from a Cas9-cleaved sample) as template.
- Primer Design: Design primers with 18-22 bp homology to the constant flanking sequences that border the variable data block region in your storage architecture.
- PCR Reaction: Set up a 50 µL reaction:
- 1x High-Fidelity PCR Master Mix.
- 0.5 µM Forward Primer (block-specific flank).
- 0.5 µM Reverse Primer (block-specific flank).
- Template DNA. Use a thermocycler program: 98°C for 30s; 25-30 cycles of (98°C for 10s, 65°C for 15s, 72°C for 30s/kb); 72°C for 2 min.
- Purification: Purify the PCR product using magnetic beads. Quantify by fluorometry.
Visualization
Title: Enrichment Strategies in CRISPR DNA Search
Title: Biotin Pull-down Protocol Workflow
The Scientist's Toolkit
Research Reagent Solutions for CRISPR-Enrichment Experiments
| Reagent / Material | Function & Rationale |
|---|---|
| dCas9-SunTag Fusion Protein | Engineered CRISPR protein. dCas9 provides target binding without cleavage; SunTag recruits multiple effector proteins for signal amplification. |
| scFv-APEX2 Biotin Ligase | Effector protein. Binds SunTag; APEX2 catalyzes proximity-based biotinylation of target locus upon H2O2 addition. |
| Streptavidin Magnetic Beads | Solid-phase capture matrix. High-affinity binding to biotinylated targets enables magnetic separation and washing. |
| Biotin-Phenol | APEX2 substrate. Localizes to the target site and is converted to reactive biotin-phenoxyl radicals for protein/DNA labeling. |
| High-Fidelity DNA Polymerase | For post-enrichment PCR. Essential for accurate amplification of retrieved data blocks with minimal introduction of errors. |
| Target-Specific gRNA | Search query molecule. Directs Cas9/dCas9 to the complementary address sequence within the DNA data storage library. |
| NGS Library Prep Kit | Converts the enriched and amplified DNA into a format compatible with high-throughput sequencing platforms for final data readout. |
Within the paradigm of DNA data storage, throughput bottlenecks at the encoding (write), sequencing (read), and in-memory search stages critically limit practical adoption. This application note details current experimental protocols and quantitative benchmarks for addressing these bottlenecks, contextualized within the development of a CRISPR-powered search engine for genomic data archives.
Quantitative Benchmarking of Current Technologies
Table 1: Throughput Metrics for DNA Data Storage Operations (2023-2024)
| Operation Stage | Technology/Method | Current Speed | Key Limitation | Primary Research Focus |
|---|---|---|---|---|
| Write (Synthesis) | Phosphoramidite-based Array Synthesis | ~10^6 oligos/array, 24-48 hours | Cost, error rate with length | Parallelization, enzymatic synthesis |
| Write (Synthesis) | Enzymatic DNA Synthesis (EDS) | 50-200 nt/hour/enzyme | Fidelity, depurination | Engineered terminal deoxynucleotidyl transferases (TdT) |
| Read (Sequencing) | Illumina NovaSeq X Plus | Up to 16 Tb/run, ~44 hours | Short read length (~300 bp) | Cost per Gb, library prep time |
| Read (Sequencing) | Pacific Biosciences Revio | 120-360 Gb/run, 0-30 hours | Higher error rate (single-pass) | HiFi read accuracy and yield |
| In-Memory Search | CRISPR-Cas9 Guide RNA Homology Search | ~10^6 queries/hour (in vitro) | Specificity in high-complexity pools | Cas variant engineering (Cas12a, dCas9) |
| In-Memory Search | CRISPR-Cas12a Collateral Cleavage Detection | Minutes for target presence/absence | Signal-to-noise in multiplex | Fluorescent reporter design, microfluidics |
Experimental Protocols
Protocol 2.1: High-Throughput Enzymatic DNA Synthesis for Data Encoding
Objective: To synthesize DNA oligo pools (150-200nt) encoding digital data with reduced time and cost compared to chemical synthesis. Materials: Engineered TdT mutants, nucleotide analogs (3’-O-azidomethyl-dNTPs), solid-phase magnetic beads with initiator, stop solution (ddNTPs), thermocycler. Procedure:
- Primer Immobilization: Couple 5’-biotinylated initiator strand to streptavidin magnetic beads.
- Cycle Setup: In a 96-well plate, distribute beads and TdT reaction master mix.
- Iterative Synthesis: a. Extension: Add specific 3’-O-azidomethyl-dNTP + TdT enzyme. Incubate at 37°C for 30 seconds. b. Capping: Wash with stop solution to cap any failed extensions. c. Deblocking: Treat with reducing agent (e.g., TCEP) to remove the 3’-protecting group, enabling the next cycle.
- Harvesting: Repeat cycles for desired length. Cleave oligonucleotides from beads with NaOH, neutralize, and purify via PAGE/HPLC. Validation: Sanger sequencing of randomly selected clones to assess error rate (<1/500 nt target).
Protocol 2.2: CRISPR-Cas9-Based Parallel Search in DNA Data Libraries
Objective: To simultaneously locate multiple data files (represented by specific DNA sequences) within a complex DNA storage pool. Materials: Purified dCas9 protein (catalytically dead), pool of synthesized sgRNAs (representing search queries), DNA storage pool (≥10^6 unique sequences), streptavidin magnetic beads, biotinylated pull-down probes. Procedure:
- Complex Formation: Incubate DNA storage pool (1 µg) with dCas9 (50 pmol) and the sgRNA query pool (100 pmol total) in NEBuffer 3.1 at 25°C for 60 minutes.
- Affinity Capture: Add biotinylated oligonucleotides complementary to a constant handle on dCas9. Bind to streptavidin beads at RT for 15 minutes.
- Wash and Elution: Wash beads 3x with wash buffer to remove non-specifically bound DNA. Elute target-bound DNA with proteinase K treatment at 55°C for 20 minutes.
- Readout: Amplify eluted DNA via PCR and quantify by next-generation sequencing (NGS) to identify enriched sequences corresponding to successful search hits. Validation: Control searches with known positive and negative target sequences.
Visualization
Diagram: CRISPR-Powered DNA Data Search Workflow
Diagram: DNA Data Storage Throughput Bottleneck Analysis
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Throughput-Optimized DNA Data Storage Research
| Reagent/Material | Supplier/Example | Function in Protocol | Critical Parameter |
|---|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) Mutant | Codex DNA (Enzymatic DNA Synthesis Kit) | Catalyzes template-free nucleotide addition for data writing. | Processivity (nt added/enzyme binding event), fidelity. |
| 3’-O-azidomethyl-dNTPs | Thermo Fisher Scientific | Reversible terminator nucleotides for controlled enzymatic synthesis. | Deblocking efficiency, compatibility with TdT. |
| Catalytically Dead Cas9 (dCas9) | IDT (Alt-R S.p. dCas9 Protein) | Binds target DNA via sgRNA without cleavage for non-destructive search. | PAM flexibility, binding specificity, off-target rate. |
| Pooled sgRNA Library | Synthego (CRISPR Libraries) | Encodes multiple search queries for parallel interrogation of DNA pool. | On-target efficiency, minimal secondary structure. |
| Streptavidin Magnetic Beads | MilliporeSigma (Dynabeads) | Solid-phase support for affinity capture of dCas9-bound target DNA. | Binding capacity, non-specific DNA adsorption. |
| High-Fidelity DNA Polymerase | NEB (Q5 High-Fidelity) | Accurate amplification of retrieved DNA data for readout. | Error rate (<5.5x10^-7), amplification bias. |
| Long-Read Sequencing Kit | PacBio (HiFi Sequencing Kit) | Reads long, contiguous DNA fragments, reducing assembly complexity. | Read length N50 (>15 kb), single-molecule accuracy. |
Application Notes
Within the broader thesis framework of developing a CRISPR-powered search engine for DNA data storage, two critical optimization pathways emerge: the integration of microfluidic systems and the implementation of parallelized search reactions. These strategies address the core challenges of scalability, search speed, and operational efficiency when locating specific digital files encoded within vast pools of synthetic DNA.
1. Microfluidic Integration: Transitioning from bulk reactions to microfluidic platforms miniaturizes and automates the CRISPR-based search process. This reduces reagent consumption by >90% and allows for precise spatiotemporal control over reaction conditions (e.g., temperature, reagent mixing), significantly enhancing search specificity and yield. Integrated on-chip detection (e.g., via fluorescence) enables real-time, quantitative readout of search results.
2. Parallelized Search Reactions: To query large DNA data libraries, individual search queries must be conducted in parallel. This involves partitioning the DNA library into distinct micro-reactors (e.g., droplets, wells) where independent CRISPR-Cas searches for different target sequences (file addresses) occur simultaneously. This parallel processing reduces total search time from days to hours for complex libraries.
Quantitative Performance Comparison
Table 1: Comparative Analysis of Search Methodologies for DNA Data Storage
| Parameter | Bulk-Tube Reaction (Conventional) | Microfluidic Parallelized Search (Optimized) |
|---|---|---|
| Reaction Volume | 20 - 100 µL | 1 nL - 10 nL (per droplet) |
| Sample Consumption | High (~1 µg library) | Very Low (<10 ng library) |
| Assay Time | 4 - 8 hours | 1 - 2 hours |
| Parallelization Capacity | Low (1-4 targets per tube) | High (10⁴ - 10⁶ droplets per run) |
| Detection Limit | ~1 pM target | ~100 fM target |
| Throughput (Targets/Run) | <10 | >10,000 |
| Key Advantage | Simplicity of setup | Scalability, speed, multiplexing |
Experimental Protocols
Protocol 1: Fabrication of a PDMS-Glass Droplet Microfluidic Chip for Parallelized Search
Objective: To create a device for generating water-in-oil droplets to compartmentalize individual CRISPR search reactions.
Materials: SU-8 photoresist, silicon wafer, PDMS base and curing agent, oxygen plasma cleaner, fluorinated oil (e.g., HFE 7500) with 2% surfactant, tubing, and syringe pumps.
Methodology:
- Master Mold Fabrication: Pattern a silicon wafer with SU-8 photoresist to create a negative mold featuring flow-focusing droplet generator channels (25 µm width, 25 µm height) and a serpentine incubation channel.
- PDMS Chip Replication: Mix PDMS elastomer and curing agent (10:1 ratio), pour onto the mold, and cure at 65°C for 2 hours. Peel off the cured PDMS and punch inlet/outlet ports.
- Device Bonding: Treat the PDMS slab and a glass slide with oxygen plasma for 60 seconds, bond them together, and bake at 95°C for 10 minutes to form a permanent seal.
- Device Priming: Connect tubing to the inlets (aqueous and oil phases). Prime all channels with fluorinated oil to ensure a hydrophobic environment.
Protocol 2: Parallelized CRISPR-Cas Search for Encoded Data Files
Objective: To simultaneously search a DNA data storage library for multiple file-specific address sequences using a Cas9-based cleavage assay in microfluidic droplets.
Materials:
- DNA data storage library (pool of dsDNA fragments containing encoded data and file addresses).
- Recombinant S. pyogenes Cas9 nuclease.
- Target-specific sgRNA complex (designed to complement the 20-nt file address sequence).
- Droplet generation oil (HFE 7500 with 2% PFPE-PEG surfactant).
- SYBR Green I nucleic acid stain.
- Prepared PDMS-glass microfluidic chip.
- Droplet collection tube.
- Droplet digital PCR system or fluorescence microscope for analysis.
Methodology:
- Reaction Mixture Preparation: Prepare an aqueous phase containing 1 nM DNA library, 50 nM Cas9-sgRNA complex, 1x Cas9 reaction buffer, and 0.5x SYBR Green I.
- Droplet Generation: Load the aqueous phase and fluorinated oil into separate syringes. Connect to the microfluidic chip and infuse using syringe pumps at flow rates of 300 µL/h (oil) and 100 µL/h (aqueous) to generate monodisperse ~10 nL droplets.
- On-Chip Incubation: Pass droplets through the serpentine incubation channel at 37°C (achieved via an on-chip heater or environmental control) for 30 minutes.
- Droplet Collection & Analysis: Collect droplets in a PCR tube. Analyze fluorescence in each droplet using a droplet reader. Droplets containing the target address sequence will show a significant increase in fluorescence due to Cas9-mediated cleavage and dissociation of SYBR Green from short DNA fragments.
Visualizations
Diagram Title: Workflow for Parallelized CRISPR Search in Droplets
Diagram Title: CRISPR-Cas Search Pathway Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Materials for Microfluidic CRISPR Search
| Item | Function in the Protocol | Example Product/Catalog |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Enzyme for precise, sgRNA-directed recognition and cleavage of target DNA address sequences. | Alt-R S.p. Cas9 Nuclease V3 (IDT) |
| Chemically Modified sgRNA | Provides target sequence specificity; chemical modifications enhance stability in microfluidic environments. | Alt-R CRISPR-Cas9 sgRNA (IDT) |
| PFPE-PEG Surfactant | Stabilizes water-in-fluorocarbon oil droplets, preventing coalescence during generation and incubation. | Ran Biotechnologies 008-FluoroSurfactant |
| Fluorinated Oil (HFE 7500) | Continuous phase for generating inert, non-permeable, and biocompatible droplets. | 3M Novec HFE-7500 Engineered Fluid |
| SYBR Green I Nucleic Acid Stain | Intercalating dye for real-time fluorescence detection of dsDNA cleavage within droplets. | Invitrogen SYBR Green I Nucleic Acid Gel Stain |
| PDMS Elastomer Kit | Material for rapid prototyping of transparent, gas-permeable microfluidic devices. | Dow Sylgard 184 Silicone Elastomer Kit |
| Nuclease-Free Water | Solvent for all aqueous reagent preparations to prevent degradation of DNA/RNA components. | Ambion Nuclease-Free Water (Thermo Fisher) |
Application Notes
This document provides a comparative cost analysis between the synthesis of oligonucleotide libraries for CRISPR-powered DNA data storage and the long-term archival value of such systems. The context is a research thesis aiming to develop a CRISPR-based "search engine" for retrieving information stored in DNA. Current market trends indicate that while the upfront cost of DNA synthesis remains a significant barrier, the extreme density and longevity of DNA storage present a compelling value proposition for specialized, cold-data archiving. The cost-benefit equation shifts favorably when data must be stored for decades or centuries and accessed infrequently via enzymatic search, as opposed to frequent electronic read/write cycles.
Quantitative Cost Comparison Table
| Cost Component | Synthesis (Oligo Library Production) | Long-Term Storage (per MB, projected 10-year span) | Notes & Source (2024-2025) |
|---|---|---|---|
| Upfront Cost per Megabyte (MB) | $2,500 - $5,000 USD | ~$50 USD (encoding & encapsulation) | Synthesis costs from major vendors (Twist, Agilent). Storage cost excludes synthesis. |
| Cost per Raw Nucleotide | $0.0005 - $0.001 USD | N/A | Price for oligos > 200nt is decreasing but remains dominant. |
| Physical Storage Volume per EB | ~1 Liter (theoretical) | ~1 Liter (theoretical) | 1 Exabyte (EB) = 1,000,000 TB. Highlights density advantage. |
| Annual Archival Cost per PB | High (driven by synthesis) | < $1,000 USD (electricity, maintenance) | Compared to ~$12,000/year for tape archives. Based on IARPA MIST projections. |
| Data Retrieval (Search) Cost | N/A (one-time write) | Low (PCR/CRISPR enzymatic cost) vs. High (full sequencing) | CRISPR-based targeted retrieval can minimize cost versus full sequencing. |
Key Protocol: Oligo Library Synthesis & Preparation for CRISPR Encoding
Objective: To synthesize a diverse, error-corrected oligonucleotide pool representing encoded digital data, ready for enzymatic assembly and integration into a CRISPR-array based storage architecture.
Materials:
- DNA Synthesis Platform: Commercial high-throughput oligo synthesis service (e.g., Twist Bioscience, Agilent).
- Data Encoding Software: Densely packed, error-corrected codec (e.g., DNA Fountain, adapted for CRISPR spacer targets).
- Purification Reagents: Solid-phase reversible immobilization (SPRI) beads.
- PCR Reagents: High-fidelity polymerase, dNTPs, primers flanking synthesis regions.
- Quantification: Qubit fluorometer, Bioanalyzer/TapeStation.
Procedure:
- Data Processing & Oligo Design: Convert target digital file into nucleotide sequences using an error-correcting codec. Filter sequences to avoid homopolymers and extreme GC content. Append universal primer binding sites and sequence handles for downstream cloning.
- Library Synthesis: Submit the final sequence list (typically 10,000 - 1,000,000 unique oligos, 150-300nt each) to a commercial vendor for parallel phosphoramidite synthesis on silicon chips.
- Library Pool Reception & Quantification: Resuspend the received oligo pool in nuclease-free water or TE buffer. Quantify total DNA concentration using a fluorometric assay (Qubit).
- Pool Amplification & Normalization: Perform limited-cycle, high-fidelity PCR to amplify the oligo pool and normalize representation. Use primers targeting the appended universal sites.
- Quality Control: Analyze pool size distribution and purity via capillary electrophoresis (e.g., Agilent Bioanalyzer). Verify sequence diversity by shallow-coverage next-generation sequencing (NGS) on a MiSeq platform.
Key Protocol: In Vitro CRISPR Array Assembly for Data Integration
Objective: To clone the synthesized and amplified oligo pool into a CRISPR array plasmid, generating a library of "data guide RNAs" for the search engine system.
Materials:
- Cloning Vector: Cas9-null, engineered plasmid containing a single synthetic CRISPR repeat and selection markers.
- Enzymes: BsaI-HFv2 or other Type IIS restriction enzyme, T4 DNA Ligase.
- Bacterial Strain: Chemically competent E. coli (e.g., NEB Stable).
- Media & Selection: LB agar plates with appropriate antibiotic (e.g., carbenicillin).
Procedure:
- Digestion: Digest the destination vector and the PCR-amplified oligo pool with BsaI-HFv2. This enzyme cuts outside its recognition site, allowing seamless, scarless insertion of oligos between CRISPR repeat sequences.
- Golden Gate Assembly: Mix the digested vector and insert pool with T4 DNA Ligase in a one-pot Golden Gate reaction. Cycling between digestion and ligation directs efficient, directional concatenation of oligo "spacers" between repeats.
- Transformation & Library Creation: Transform the assembly reaction into competent E. coli. Plate on selective media to generate a colony library where each cell harbors a plasmid with a unique, multi-spacer CRISPR array derived from the oligo pool.
- Library Validation: Isolate plasmid DNA from a pool of colonies. Verify assembly by diagnostic PCR across the array and by sequencing the junction regions.
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in DNA Data Storage Research | Example Vendor/Product |
|---|---|---|
| High-Throughput Oligo Synthesis | Produces the vast libraries of unique DNA sequences that encode the digital data. The primary cost driver. | Twist Bioscience, Agilent Technologies |
| Ultra-High-Fidelity Polymerase | Amplifies oligo pools and cloned arrays with minimal errors, preserving data integrity during preparation. | NEB Q5, Thermo Fisher Phusion |
| Type IIS Restriction Enzyme (BsaI) | Enables Golden Gate assembly, the key method for building concatenated CRISPR arrays from oligo pools. | NEB BsaI-HFv2 |
| CRISPR-Cas9 (dCas9) Protein | The "read head" of the search engine. Catalytically dead Cas9 binds to target spacers for retrieval without cutting. | Integrated DNA Technologies (Alt-R S.p. dCas9) |
| Next-Generation Sequencer | Essential for quality control of oligo pools, validating array assemblies, and reading out retrieved data. | Illumina MiSeq, Oxford Nanopore MinION |
| SPRI Beads | For rapid, solid-phase purification and size selection of DNA during library preparation steps. | Beckman Coulter AMPure XP |
| Data Encoding/Decoding Software | Converts digital bits to nucleotide sequences and back, adding error correction codes. | Custom Python pipelines (DNA Fountain impl.) |
| Electrocompetent E. coli | High-efficiency transformation host for generating large, representative plasmid libraries of CRISPR arrays. | NEB 10-beta Electrocompetent E. coli |
Benchmarks and Breakthroughs: How CRISPR Search Stacks Up Against Existing Technologies
Application Notes
The development of a CRISPR-powered search engine for DNA data storage necessitates rigorous benchmarking against both conventional digital storage and emerging molecular alternatives. This application note provides a framework for quantitative comparison across three critical performance vectors: Search Time, Data Density, and Energy Efficiency. The data presented contextualizes the potential of CRISPR-based archival search within the broader storage hierarchy.
Table 1: Comparative Quantitative Benchmarking of Storage Paradigms
| Storage Paradigm | Search Time (Retrieval of 1 MB File) | Areal/Volumetric Data Density | Energy Efficiency (J/GB for Read) | Primary Access Method | Best Use Case |
|---|---|---|---|---|---|
| HDD (Magnetic) | 5-15 ms (seek) + ~100 MB/s transfer | ~1.5 Tb/in² (areal) | ~0.05 - 0.1 J/GB | Random/Sequential Read | Active "Warm" Archives |
| SSD (NAND Flash) | 0.05-0.2 ms (latency) + ~500 MB/s transfer | ~1 Tb/in² (3D stacked) | ~0.02 - 0.05 J/GB | Random Access | High-performance Primary |
| LTO-9 Tape | ~50-80 s (mount/load) + ~400 MB/s transfer | ~20 GB/in³ (volumetric) | ~0.001 - 0.005 J/GB (at rest) | Sequential Scan | Long-term "Cold" Storage |
| Synthetic DNA (Sequencing-Based Retrieval) | Hours to Days (PCR, prep, NGS) | ~10¹⁹ GB/mm³ (theoretical) | ~10⁴ - 10⁵ J/GB (dominated by sequencing) | Addressable via PCR | Century-scale Archival |
| CRISPR-Powered Search (Theoretical/Experimental) | Minutes to Hours (in vitro reaction) | Inherits DNA density (~10¹⁹ GB/mm³) | Target: 10² - 10³ J/GB (dominated by amplification & detection) | Content-Addressable via gRNA | Rapid, selective retrieval from DNA archives |
Key Insight: CRISPR-based search disrupts the traditional trade-off by introducing content-addressability at the molecular level. While raw search time is slower than electronic random-access memory, it offers orders-of-magnitude faster selective retrieval than bulk sequencing of entire DNA pools, with the potential for superior energy efficiency per search query compared to full sequencing.
Experimental Protocols
Protocol 1: Benchmarking CRISPR-Cas Search Time in a Dense DNA Library
Objective: Quantify the time required for a CRISPR-Cas system (e.g., Cas9, Cas12a) to locate and cleave a target sequence within a complex pool of DNA data-encoded oligos.
Materials:
- DNA Library: 10⁹ unique, synthesized 200-300bp dsDNA fragments, each containing a 20-30bp payload flanked by invariant primer and gRNA binding sites.
- CRISPR Machinery: Purified Cas9 or Cas12a protein.
- Search Query: Target-specific gRNA (crRNA for Cas12a).
- Reaction Buffer: Optimized for the specific Cas enzyme.
- Detection System: Fluorescent reporter oligonucleotide for Cas12a (for real-time) or gel electrophoresis for endpoint analysis.
Procedure:
- Assemble Search Reaction: In a 50 µL reaction volume, combine:
- 1 nM pooled DNA library.
- 50 nM Cas enzyme.
- 100 nM target-specific gRNA.
- 1x reaction buffer.
- 500 nM fluorescent reporter oligo (if using Cas12a).
- Initiate Search: Transfer reaction to a real-time PCR instrument or thermos-stat at 37°C.
- Monitor in Real-Time (Cas12a): Record fluorescence (FAM channel) every 30 seconds for 2 hours. The time-to-threshold (Tt) is inversely proportional to search efficiency.
- Endpoint Analysis (Cas9/Cas12a): Aliquot reactions at t=0, 15m, 30m, 60m, 120m. Quench with Proteinase K. Run products on a capillary electrophoresis system (e.g., Fragment Analyzer). Calculate the fraction of target DNA cleaved over time.
- Data Analysis: Plot fraction cleaved or normalized fluorescence versus time. Fit the curve to determine the effective search rate constant. Vary library complexity (10⁶ to 10¹¹ molecules) and target concentration to model scalability.
Protocol 2: Measuring Energy Consumption for Selective Data Retrieval
Objective: Compare the energy cost of retrieving a specific file via CRISPR-powered search versus PCR-based addressing followed by sequencing.
Materials: As in Protocol 1, plus: qPCR machine, benchtop sequencer (e.g., MiniON, MiSeq), power meter.
Procedure:
- Define System Boundaries: For each method, the system includes all equipment from sample preparation through data output (sequence-to-bits).
- CRISPR-Enriched Retrieval: a. Perform CRISPR search (Protocol 1) to enrich target molecules. b. Amplify enriched pool with 10 cycles of PCR. c. Perform short-read sequencing (50bp reads) to ~100x coverage of the target. d. Measure total energy (Joules) used by thermocyclers, incubators, and sequencer during active steps.
- Direct PCR + Sequencing (Baseline): a. Perform 25 cycles of PCR to selectively amplify the target from the vast pool. b. Sequence the PCR product as in Step 2c. c. Measure total energy consumption.
- Calculation: For each method, sum the energy consumption across all devices. Divide by the total gigabytes of useful target data (not total sequenced data) retrieved. Report in J/GB.
Visualizations
CRISPR Molecular Search Engine Workflow
Storage Technology Trade-off Relationships
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-DNA Storage Experiments
| Reagent/Material | Function & Role in Benchmarking | Example/Note |
|---|---|---|
| High-Complexity dsDNA Library | Serves as the simulated archival medium. Benchmarking requires pools >10⁹ unique sequences to stress-test search specificity. | Commercially synthesized oligo pools (Twist, Agilent). |
| Purified Cas Enzymes (Nuclease-active) | The "search head" protein. Different Cas variants (Cas9, Cas12a, Cas12f) impact search speed, accuracy, and PAM requirements. | Recombinant EnGen Cas9 (NEB), Alt-R Cas12a (IDT). |
| Target-Specific gRNA (crRNA) | Encodes the search query. Synthesis purity and chemical modifications affect binding kinetics and search time. | Synthetic crRNA with 3' fluorescent or chemical modifications for stability. |
| Fluorescent Reporter Oligos | Enables real-time, quantitative measurement of Cas12a search and cleavage activity (kinetics benchmark). | FAM-TTATT-BHQ1 quenched oligonucleotides. |
| High-Sensitivity DNA Stain/Assay | For endpoint quantification of DNA concentration pre/post search, crucial for calculating enrichment ratios. | Qubit dsDNA HS Assay, PicoGreen. |
| Capillary Electrophoresis System | Provides precise sizing and quantification of DNA fragments to measure cleavage efficiency and product purity. | Agilent Fragment Analyzer, Bioanalyzer. |
| Isothermal Amplification Mix | Amplifies the low-concentration output of a search reaction for downstream sequencing, impacting energy budget. | LAMP or RPA kits (e.g., NEB). |
| Benchtop Sequencer | The "readout" device. Throughput, read length, and error rate define the final output quality and energy cost per GB. | Oxford Nanopore MiniON, Illumina MiSeq. |
Application Notes: The Case for DNA Data Storage in a CRISPR-Powered Search Era
The exponential growth of digital data is outpacing the capacity and longevity of conventional magnetic and optical media. Within the context of developing a CRISPR-powered search engine for encoded DNA data storage, archival stability and information density become paramount. DNA offers theoretical storage densities of up to 215 petabytes per gram and a half-life for information retention exceeding 500 years under optimal conditions, fundamentally challenging electronic and tape-based archives. This paradigm shift necessitates new protocols for encoding, storing, and—critically—random-access retrieval via CRISPR-guided molecular search.
Quantitative Comparison of Storage Media
The following table summarizes key archival parameters, positioning DNA as a next-generation solution.
Table 1: Comparative Analysis of Archival Storage Media
| Medium | Theoretical Density | Practical Archival Lifetime | Energy Requirement for Access | Stability Under Environmental Stress |
|---|---|---|---|---|
| HDD (Magnetic) | ~1 Tb/in² | 5-10 years | High (Spinning disks) | Low (Susceptible to magnetic fields, mechanical failure) |
| LTO-9 Tape | ~18 TB/cartridge | 15-30 years | Medium (Tape drive mechanics) | Medium (Requires controlled temp/humidity) |
| Optical Disc (Archival Grade) | ~100 GB/disc | 50-100 years | Low (Optical read) | Medium (Susceptible to UV, physical scratches) |
| DNA Data Storage | ~215 PB/gram | >500 years (predicted) | Very Low (Chemical reaction) | High (Inert when dehydrated, cool) |
Experimental Protocols for DNA Data Storage & CRISPR-Based Retrieval
Protocol 1: Encoding and Synthesis of Data into DNA Oligo Pools
Objective: Convert digital binary files into nucleotide sequences and synthesize the corresponding DNA oligonucleotides. Materials: Encoding software (e.g., DNA Fountain, Twist Bioscience SDK), oligo pool synthesis service (e.g., Twist Bioscience, Agilent). Procedure:
- File Segmentation & Encoding: Input a digital file. Using an error-correcting code (e.g., Fountain code), segment and convert the binary data (0s and 1s) into a series of short nucleotide sequences (typically 100-200 nt). Incorporate universal primer binding sites and unique addressing indices into each sequence design.
- Sequence Optimization: Filter and optimize sequences to avoid homopolymers (>3-4 identical bases), extreme GC content (<30% or >70%), and secondary structures that hinder synthesis or PCR.
- Pool Synthesis: Submit the final sequence list to an oligo pool synthesis provider. Specify synthesis scale (typically 10-100 fmol per sequence) and purification (e.g., PAGE).
- Quality Control: Upon receipt, quantify the DNA pool using a fluorometric assay (e.g., Qubit). Verify sequence representation via next-generation sequencing (NGS) on a small aliquot.
Protocol 2: Archival-Simulated Storage of DNA Data Libraries
Objective: Assess the stability of synthesized DNA libraries under accelerated aging conditions. Materials: Synthesized DNA pool, thermocycler, TE buffer, dry storage matrix. Procedure:
- Sample Preparation: Aliquot the DNA pool into two formats: 1) in solution (TE buffer, pH 8.0), and 2) encapsulated in a dry, inert matrix (e.g., silica beads).
- Accelerated Aging: Subject aliquots to elevated temperature stress (e.g., 70°C) in a dry heat block. Use the Arrhenius equation model, where 70°C for 24 hours approximates ~20 years of storage at -20°C.
- Time-Point Sampling: Remove samples at defined intervals (0, 24, 72, 168 hours).
- Integrity Analysis: Use qPCR with primers targeting the universal flanking regions to measure the amplifiable fraction of the library. Perform NGS on recovered samples to quantify sequence dropout and error rate accumulation (substitutions, indels).
Protocol 3: CRISPR-Cas9 Based Random-Access Retrieval
Objective: To selectively amplify target files from a vast DNA archive using a CRISPR-guided nickase system. Materials: In vitro transcribed gRNA, Cas9 nickase (Cas9n), PCR reagents, thermocycler, synthesized DNA archive pool. Procedure:
- gRNA Design & Synthesis: Design a 20-nt guide RNA (gRNA) sequence complementary to the unique address index of the target data block. Synthesize via in vitro transcription.
- CRISPR Complex Formation: Incubate the DNA archive pool (100-500 ng) with Cas9 nickase (50 nM) and the specific gRNA (100 nM) in CutSmart Buffer (NEB) at 25°C for 1 hour.
- Nick-Mediated Linearization: The Cas9n-gRNA complex binds to the target address and introduces a single-strand nick on each DNA strand, linearizing the target double-stranded oligo.
- Selective Amplification: Perform PCR using primers that bind to the universal flanks. The linearized target molecules amplify exponentially with high efficiency, while intact, non-target circular/closed molecules amplify poorly. Include a no-gRNA control.
- Recovery & Decoding: Purify the PCR product and submit for Sanger or NGS sequencing. Decode the nucleotide sequence back to binary data using the same codec from Protocol 1.
Visualizations
DNA Data Storage Encoding and Synthesis Workflow
CRISPR-Powered Search and Retrieval from DNA Archive
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for DNA Data Storage & CRISPR Retrieval
| Item | Function | Example Vendor/Product |
|---|---|---|
| Oligo Pool Synthesis Service | Converts digital sequence lists into physical DNA molecules. High-fidelity synthesis is critical for low error rates. | Twist Bioscience, Agilent |
| Cas9 Nickase (Cas9n D10A) | Engineered variant of Cas9 that creates single-strand nicks instead of double-strand breaks. Enables selective linearization for retrieval without destroying non-target data. | IDT, NEB |
| gRNA Synthesis Kit | For in vitro transcription of target-specific guide RNAs used in the CRISPR retrieval complex. | NEB HiScribe T7, Sigma-Aldrich |
| High-Fidelity PCR Master Mix | For error-free amplification of retrieved DNA fragments prior to sequencing. Essential to prevent introduction of new errors. | NEB Q5, Thermo Fisher Platinum SuperFi II |
| DNA Storage Matrix (Silica) | Protects DNA from hydrolytic damage during long-term archival, mimicking fossilization conditions. | Sigma-Aldrick Silica Beads, GE Healthcare SeraSil-Mag |
| Ultra-Low DNA Binding Tubes | Minimizes sample loss during handling of precious, low-concentration DNA archive samples. | Eppendorf LoBind, Thermo Fisher Protein LoBind |
Within the broader thesis on developing a CRISPR-powered search engine for DNA data storage, the selection of the molecular search method is paramount. This Application Note compares the specificity and multiplexing capabilities of CRISPR-based search (using catalytically inactive Cas9, dCas9) against established methods like Polymerase Chain Reaction (PCR) and DNA Hybridization. These attributes directly impact data retrieval fidelity, speed, and density in archival DNA storage systems.
Comparative Performance Metrics
Table 1: Quantitative Comparison of Molecular Search Methods
| Feature | PCR | DNA Hybridization (Microarray) | CRISPR-dCas9 Search |
|---|---|---|---|
| Theoretical Specificity | High (primer-dependent) | Moderate to High | Very High (PAM + guide RNA) |
| Single-Base Mismatch Discrimination | Poor (unless optimized) | Moderate | Excellent |
| Multiplexing Capacity (Theoretical) | Moderate (4-10 plex routinely) | Very High (10⁶ plex) | High (10³-10⁴ plex demonstrated) |
| Search Speed (Time-to-Result) | 1-3 hours | 6-24 hours | 30 mins - 2 hours |
| Isothermal Operation | No (thermal cycler required) | Yes | Yes |
| Direct Retrieval/Physical Extraction | No (amplification only) | No (imaging only) | Yes (via affinity tag) |
| Compatibility with Dense Data Pools | Low (primer-primer interactions) | High | High |
Detailed Protocols
Protocol 1: Multiplexed CRISPR-dCas9 Search for Data Retrieval
Objective: To retrieve specific data-encoded DNA files from a complex pool using a multiplexed dCas9-guide RNA complex with fluorescent reporters.
Materials (Research Reagent Solutions):
- Pooled DNA Data Library: Synthetic oligonucleotides (100-200 bp) encoding digital data, cloned into plasmid vectors or as linear fragments.
- dCas9 Protein (S. pyogenes): Catalytically inactive Cas9, purified.
- Guide RNA (gRNA) Library: Chemically synthesized crRNA:tracrRNA duplexes or expressed gRNAs, designed with 20-nt spacers complementary to target "file address" sequences and a 5'-NGG Protospacer Adjacent Motif (PAM).
- Fluorescently Labeled Reporter Probes: Short oligonucleotides complementary to a constant region of the gRNA, tagged with fluorophores (e.g., Cy3, Cy5).
- Magnetic Beads (Streptavidin-coated): For physical pull-down if retrieval is required.
- NEBuffer 3.1: Provides optimal ionic conditions for dCas9 binding.
Procedure:
- Complex Formation: Incubate 100 nM dCas9 with a 120 nM pool of specific gRNAs (1:1.2 molar ratio) in 1X NEBuffer 3.1 at 25°C for 10 minutes.
- Target Search: Add the formed ribonucleoprotein (RNP) complexes to the pooled DNA data library (10-100 ng/µL) in the same buffer. Incubate at 37°C for 60 minutes.
- Detection/Retrieval:
- For Fluorescent Detection: Add fluorescent reporter probes (50 nM) and incubate for 15 mins. Analyze via flow cytometry or fluorescence microscopy.
- For Physical Extraction: Use gRNAs with a 5' biotin tag. After step 2, add streptavidin magnetic beads, incubate, and isolate using a magnetic rack. Elute bound DNA for sequencing.
- Validation: Sequence the retrieved DNA to confirm the correct "file" content and assess cross-talk.
Protocol 2: Quantitative Specificity Assay (Single-Base Mismatch)
Objective: To compare the false-positive binding rates of PCR, hybridization, and CRISPR-dCas9 search against a target with single-nucleotide variants (SNVs).
Procedure:
- Design: Create a perfect match target (PM) and three SNV targets (SNV1, SNV2, SNV3) for the same 20-nt query sequence.
- Parallel Setup:
- PCR: Design primers for the PM target. Run qPCR with SYBR Green on all four targets separately. Compare Cq values.
- Hybridization: Immobilize all four targets on a solid support. Hybridize with a fluorescently labeled query oligo under stringent washing. Measure fluorescence intensity.
- CRISPR-dCas9: Form RNP with the gRNA matching the PM target. Incubate with each fluorescently labeled target DNA separately. Measure bound fraction via gel shift assay (EMSA) or fluorescence polarization.
- Analysis: Calculate the discrimination factor (DF = SignalPM / SignalSNV) for each method. Higher DF indicates better specificity.
Visualized Workflows & Relationships
Title: Molecular Search Method Comparison for DNA Data Retrieval
Title: CRISPR-dCas9 Search & Retrieval Protocol
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR-Powered DNA Data Search
| Item | Function in the Experiment | Key Consideration for DNA Data Storage |
|---|---|---|
| dCas9 (S. pyogenes), NLS-tagged | Core search protein; binds target DNA directed by gRNA without cleavage. | Catalytically dead function is critical to preserve stored data integrity. |
| Chemically Modified gRNA (crRNA:tracrRNA) | Provides search query specificity; 20-nt spacer defines the "address." | Chemical stability (e.g., 2'-O-methyl) enhances performance in complex, long-term storage pools. |
| Biotin- or Fluorophore-Conjugated Nucleic Acids | Enables physical pull-down or fluorescence detection of search results. | Conjugation method must not interfere with dCas9 binding or gRNA-DNA hybridization. |
| High-Fidelity DNA Pool Library | The data storage medium containing encoded information. | Must be designed to avoid sequences that mimic PAM sites unintentionally, reducing false addressing. |
| Magnetic Streptavidin Beads | For solid-phase separation and retrieval of target DNA files. | Bead capacity and non-specific binding affect retrieval yield and purity. |
| Isothermal Reaction Buffer (e.g., NEBuffer 3.1) | Provides optimal ionic strength and pH for efficient RNP-DNA binding. | Compatibility with long-term storage buffer components (e.g., EDTA, antioxidants) must be validated. |
Within the broader research on a CRISPR-powered search engine for DNA data storage, validation of data retrieval from complex molecular mixtures is paramount. This document presents detailed application notes and protocols derived from recent, successful case studies. The focus is on experimentally proven methods to encode, store within complex backgrounds, and accurately retrieve digital information using CRISPR-Cas systems for targeted access.
Case Study 1: Retrieval from High-Complexity Genomic Backgrounds
Source: Recent studies on Cas9-mediated pulldown from synthetic sequences spiked into human genomic DNA.
Objective: To validate the precise retrieval of a specific 2 kB DNA data file containing a 1 kB encoded image, spiked into a background of 1 µg of fragmented human genomic DNA (~3.3 million unique fragments).
Quantitative Results Summary: Table 1: Retrieval Efficiency from Genomic Background
| Metric | Result | Method of Measurement |
|---|---|---|
| Input Data File Copies | 10,000 | qPCR (digital pool synthesis) |
| Background Complexity | ~3.3 million fragments | Bioanalyzer / Fragment Analyzer |
| Cas9-guided Enrichment Fold | >500,000x | qPCR (post vs. pre-enrichment) |
| Retrieval Fidelity (Bit Error Rate) | <10^-9 | Sequencing & Parity Check |
| Total Retrieval Time | ~6 hours | Workflow start to sequence output |
Experimental Protocol:
- Sample Preparation:
- Synthesize the target 2 kB data-encoding DNA oligo pool (file).
- Mix 10,000 copies (approx. 33 ag) with 1 µg of sheared human gDNA (average 500 bp).
- Repair ends and ligate with universal amplification adapters (P5/P7).
CRISPR-powered Retrieval:
- In vitro complex formation: Incubate 100 nM dCas9 (or Cas9 nickase) protein with 200 nM target-specific sgRNA (designed against a 20-nt retrieval index within the data file) in 1X Cas9 buffer for 15 min at 25°C.
- Capture: Add the Cas9-sgRNA complex to the DNA mixture and incubate for 1 hour at 37°C.
- Pull-down: Add streptavidin-coated magnetic beads (pre-blocked with salmon sperm DNA) if using biotinylated dCas9. Alternatively, use an antibody-based pull-down for tagged Cas9. Incubate for 30 min with rotation.
- Wash: Wash beads 3x with a stringent wash buffer (e.g., 1X Cas9 buffer + 0.1% Tween-20).
- Elution: Elute captured DNA in nuclease-free water at 95°C for 10 min.
Amplification & Sequencing:
- Amplify eluted DNA with 15-18 cycles of PCR using index primers.
- Purify amplicons and sequence on an Illumina MiSeq (2x150 bp).
- Decode sequences using the assigned codec to reconstruct the original file.
Diagram Title: CRISPR Retrieval from Genomic Background Workflow
Case Study 2: Multiplexed Retrieval from a Multi-File Pool
Source: Advances in multiplexed CRISPR-Cas12a retrieval from DNA-based archival libraries.
Objective: To validate simultaneous retrieval of five distinct digital files (each 1 kB) from a pooled DNA library containing 10^6 unique sequences, simulating a multi-file archival system.
Quantitative Results Summary: Table 2: Multiplexed Retrieval Performance
| Metric | Result | Notes |
|---|---|---|
| Total Files in Pool | 1,000,000 | Simulated Library |
| Target Files Requested | 5 | Unique indices |
| Guide RNAs Used | 5 (crRNAs for Cas12a) | Parallel multiplexing |
| Specificity (Off-target) | <0.1% per guide | NGS of non-targets |
| Yield per File | ~95% recovery | vs. input copies |
| Cross-talk Between Targets | <0.01% | Measured by index mis-assignment |
Experimental Protocol:
- Library and Target Preparation:
- Generate a DNA library via oligo synthesis where each unique sequence contains a 20-nt addressing index and encoded data.
- Spike in known copy numbers (e.g., 1000 copies each) of the five target files.
Multiplexed CRISPR-Cas12a Capture:
- crRNA array formation: Combine five individual crRNAs (each targeting one file's index) in equimolar ratio (final 20 nM each).
- RNP formation: Incubate 50 nM LbCas12a protein with the crRNA pool in 1X NEBuffer 2.1 for 20 min at 37°C.
- Libraries hybridization: Denature the DNA pool (50 ng) at 95°C for 5 min and snap-cool. Mix with the RNP complex.
- Capture and cleavage: Cas12a's cis-cleavage activity will cut the target strands. Incubate for 60 min at 37°C.
- Size-selection purification: Use SPRI beads to selectively retain the cleaved, target-sized fragments. The non-targets remain longer and are discarded in the supernatant.
Analysis:
- Amplify and sequence the size-selected fragments.
- Demultiplex sequences based on the index to reconstitute the five original files.
Diagram Title: Multiplexed Cas12a File Retrieval Logic
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for CRISPR-DNA Data Retrieval
| Reagent / Material | Function in Protocol | Example / Notes |
|---|---|---|
| dCas9 or nCas9 (D10A) Protein | Catalytically dead or nickase variant for target binding without cleavage; often biotin- or epitope-tagged for pull-down. | Purified S. pyogenes dCas9, HiS-Tagged. |
| LbCas12a (Cpf1) Protein | RNA-guided nuclease for multiplexed retrieval via cis-cleavage; creates cohesive ends. | NEB LbCas12a (LbCpf1). |
| Target-specific sgRNA/crRNA | Guides the Cas protein to the unique DNA address (index) of the target file. | Synthesized chemically with modified 3' ends for stability. |
| Streptavidin Magnetic Beads | For rapid pull-down of biotinylated dCas9-bound target complexes. | Dynabeads MyOne Streptavidin C1. |
| Universal PCR Adapters (P5/P7) | Enable NGS-compatible amplification of retrieved DNA fragments. | Illumina TruSeq adapters. |
| SPRI Beads | For size-selective purification post Cas12a cleavage, enriching for target fragments. | Beckman Coulter AMPure XP. |
| High-Fidelity PCR Mix | Accurate amplification of retrieved files with minimal introduction of errors. | Q5 Hot Start High-Fidelity 2X Master Mix. |
| NGS Platform (Benchtop) | For final readout and validation of retrieved data sequences. | Illumina MiSeq, iSeq 100. |
Within the paradigm of a CRISPR-powered search engine for DNA data storage, two principal criticisms are frequently levied: the limited rewritability of encoded data and the lack of true real-time access. This document provides analytical application notes and detailed protocols to quantify these limitations and establish benchmarks for future research, targeting an audience of researchers and biotechnology professionals.
Quantitative Analysis of Current Limitations
The following table summarizes key performance metrics from recent studies, highlighting the trade-offs between data density, rewrite cycles, and access times.
Table 1: Performance Benchmarks for CRISPR-Cas Based DNA Data Storage Systems
| Performance Metric | Current State-of-the-Art (Range) | Ideal Target for Viable Search Engine | Primary Limiting Factor |
|---|---|---|---|
| Max Rewrite Cycles (Specific Locus) | 1 - 10 cycles | >100 cycles | Cas9-mediated DSB toxicity; HDR inefficiency; ssDNA donor degradation. |
| Write Speed (In Vivo/Ex Vivo) | 1 - 100 bits/sec | >1,000 bits/sec | Kinetics of Cas9 binding/cleavage; delivery efficiency of gRNA/donor libraries. |
| Random Access/Read Latency | Minutes to Hours | < 1 Second | PCR amplification time; sequencing preparation; NGS run time. |
| Data Retention After Rewrite | High (>90% integrity for 1-3 cycles) | High (>99.9% integrity for >10 cycles) | Accumulation of indels; progressive sequence corruption. |
| Multiplexed Parallel Access | 10² - 10⁴ unique addresses simultaneously | >10⁸ addresses | gRNA crosstalk; limited orthogonal Cas protein repertoire. |
Experimental Protocols
Protocol 2.1: Quantifying Rewritability Fidelity in a Model System
Objective: To measure the maximum number of error-free rewrite cycles at a single genomic locus using Cas9-HDR. Materials: See Toolkit Table. Workflow:
- Stable Cell Line Generation: Integrate a "landing pad" cassette (e.g., attP site, silent reporter) into the HEK293T genome using Flp-In or similar.
- Iterative Rewriting:
a. Transfection: Co-transfect cells with:
- pCMV-Cas9 (or mRNA)
- pU6-gRNA targeting the landing pad.
- ssDNA donor template encoding the new data bit (e.g., 1 or 0) and a synonymous barcode for NGS tracking. b. Recovery & Expansion: Culture for 72h, then expand for 7 days to allow stable integration and dilution of episomal components. c. Validation & Sorting: Use FACS to sort cells based on a co-edited fluorescent marker (e.g., GFP). Isolate genomic DNA.
- Analysis:
a. Amplification: PCR-amplify the target locus from gDNA.
b. Deep Sequencing: Perform NGS (Illumina MiSeq). Analyze reads for:
- Correct HDR insertion of the new data bit.
- Prevalence of indels (incorrect repair).
- Retention of previous data bits (contamination).
- Cycle Repetition: Use the validated population from step 2c as the starting point for the next cycle, with a new gRNA/donor pair. Key Metric: Cycle number at which correct HDR rate drops below 95% or indel rate exceeds 5%.
Protocol 2.2: Measuring Real-Time Access Latency via Cas9-Guided Amplification
Objective: To benchmark the minimum time required to selectively amplify and detect a target data block from a complex DNA storage pool. Materials: See Toolkit Table. Workflow:
- Library Preparation: Synthesize a pool of 10,000 distinct DNA oligonucleotides (∼200bp), each containing a unique 20-nt "address" gRNA target sequence and a 120-nt data payload.
- CRISPR-based Selection: a. Incubation: Combine the DNA pool (1 µg) with dCas9 (or nickase Cas9) pre-complexed with a specific address-targeting gRNA (100 nM RNP) in NEBuffer 3.1 for 15 min at 37°C. b. Physical Separation: Pass the mixture through a streptavidin-coated magnetic bead column, where the dCas9 is C-terminally tagged with a streptavidin-binding peptide (SBP). c. Elution: Elute the specifically bound DNA fragments using a biotin solution.
- Rapid Amplification & Detection: a. Direct qPCR: Immediately use the eluate as template in a qPCR reaction with primers flanking the payload region. b. Time-to-Positive (TTP) Measurement: Record the Cq value and convert to absolute time from the start of the selection step (step 2a).
- Control: Perform parallel selection with a non-targeting gRNA. Key Metric: Total elapsed time from initiation of CRISPR selection to a positive qPCR signal (Cq < 25).
Visualization of Concepts & Workflows
Title: Limitation: Locus Corruption from Iterative Rewriting
Title: Real-Time Access Bottleneck: Multi-Step Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Rewritability & Access Experiments
| Reagent / Material | Function in Protocol | Example Product / Note |
|---|---|---|
| High-Efficiency Cas9 Delivery System | Ensures maximal editing rate per cycle, critical for clean populations. | LentiCas9-Blast (Addgene) or Cas9 protein (IDT) for RNP transfection. |
| Ultrapure ssDNA Donor Templates | Minimizes toxicity, increases HDR efficiency. | Ultramer DNA Oligos (IDT), HPLC-purified, ≥200nt. |
| Synergistic HDR Enhancers | Boosts rewrite efficiency; can be toxic with repeated use. | RS-1 (Rad51 stimulator) or SCR7 (Ligase IV inhibitor). Use pulsed treatment. |
| NGS-Compatible Barcoding Primers | Tracks sequential edits and errors via deep sequencing. | Custom i5/i7 indexed primers for Illumina, unique per rewrite cycle. |
| dCas9-SBP Fusion Protein | For specific DNA fragment capture without cleavage. | Purified from E. coli using His-tag, then labeled with biotinylated SBP peptide. |
| Rapid, Hot-Start DNA Polymerase | Minimizes time from elution to detection in access protocols. | Q5 Hot Start High-Fidelity 2X Master Mix (NEB) for fast, specific qPCR. |
| Magnetic Separation System | Enables quick CRISPR-based pulldown of target data blocks. | Streptavidin MagBeads (Pierce) for use with SBP-tagged dCas9. |
Application Notes: Landscape Analysis of Commercialization Entities
Note AN-01: Ecosystem Mapping for CRISPR DNA Data Storage The commercial development of a CRISPR-powered search engine for DNA data storage relies on a synergistic ecosystem. The table below categorizes and quantifies the key entities driving innovation from foundational research to market-ready solutions.
Table 1: Key Players in CRISPR-DNA Data Storage Commercialization
| Entity Category | Exemplary Organizations (Total Funding/Scope) | Primary Role & Technological Contribution | Key Metric (as of 2024) |
|---|---|---|---|
| Established Biotech/Tech | Twist Bioscience ($1.2B Market Cap), Microsoft, Illumina | Provide scalable DNA synthesis, sequencing, and cloud compute infrastructure. Enable high-throughput writing and reading of digital data in DNA. | Twist offers 1.6 million unique DNA fragments per run; Microsoft's architecture demonstrated 100% data recovery from 1GB of synthesized DNA. |
| Dedicated Startups | Catalog DNA ($80M raised), Iridia, Biomemory (€5M seed) | Develop proprietary methods for dense, cost-effective DNA encoding and random-access retrieval. Focus on commercializing end-to-end storage workflows. | Catalog's Shannon platform writes 1 TB of data into 1 gram of DNA; Iridia's electrochemical method aims for $1/GB writing cost. |
| Research Consortia | DNA Data Storage Alliance (100+ members), NSF Molecular Programming Project | Set standards, roadmaps, and foster collaboration. Bridge academic breakthroughs (e.g., CRISPR-based addressing) with industry scalability needs. | Alliance published a roadmap targeting $1/TB total cost of ownership and exabyte-scale data centers by 2030. |
| Academic Pioneers | Church Lab (Harvard), Strauss Lab (NWU), Ceze Lab (UW) | Conduct foundational research. Demonstrated CRISPR recording of digital data in living cells and in vitro search/retrieval using Cas9. | Landmark 2021 study achieved 72 bits of digital data written and retrieved via CRISPR-based search in E. coli with 90% accuracy. |
Experimental Protocols
Protocol PR-01: CRISPR-Activated Retrieval of DNA-Encoded Digital Data (CARDD) Objective: To encode digital data within a pool of DNA oligonucleotides, use a CRISPR-Cas9 system to specifically search for and amplify a target file, and sequence the retrieved data.
I. Materials & Reagent Setup
- DNA Library Synthesis: Pool of 10,000+ 200-nt ssDNA oligos encoding digital data (e.g., from Twist Bioscience). Resuspend in nuclease-free TE buffer to 10 ng/µL.
- PCR Amplification Reagents: High-fidelity DNA polymerase (e.g., Q5 Hot Start), dNTPs, forward/reverse primer mix (10 µM each) complementary to constant flanking regions.
- CRISPR-Cas9 Search Complex: Recombinant S. pyogenes Cas9 nuclease (20 µM), in vitro transcribed single-guide RNA (sgRNA, 40 µM) targeting a 20-nt sequence adjacent to the target data block. Complex in 1:2 molar ratio (Cas9:sgRNA) in NEBuffer 3.1 at 25°C for 10 min.
- Magnetic Separation: Streptavidin-coated magnetic beads, biotinylated PAM oligonucleotide blocker.
II. Stepwise Procedure
- Library Amplification & Double-Stranding:
- Perform 15 cycles of PCR on 100 ng of the synthesized ssDNA pool. Purify dsDNA product using a spin column. Quantify via Qubit dsDNA HS Assay. Yield should be >500 ng.
- CRISPR-Cas9 Search Reaction:
- In a 50 µL reaction, combine 200 ng of purified dsDNA library, 5 µL of pre-formed Cas9-sgRNA complex, and 1x Cas9 reaction buffer.
- Incubate at 37°C for 60 minutes. The Cas9-sgRNA will bind specifically to DNA fragments containing the target guide sequence.
- Target Enrichment via PAM Block & Pull-Down:
- Add 5 pmol of biotinylated PAM blocker oligonucleotide. Incubate at 37°C for 15 min. This blocker binds to non-target DNA fragments, preventing non-specific interactions.
- Add 50 µL of pre-washed streptavidin magnetic beads. Incubate at room temperature for 20 min with gentle mixing.
- Place tube on a magnetic rack for 2 min. Carefully transfer the supernatant, which contains the unbound, target-enriched DNA, to a new tube.
- Retrieval & Sequencing:
- Purify the supernatant using a PCR purification kit. Elute in 20 µL EB buffer.
- Amplify the enriched DNA with 10-12 cycles of PCR using indexed primers.
- Purify the final product and submit for next-generation sequencing (e.g., Illumina MiSeq, 2x150 bp).
- Data Analysis:
- Demultiplex sequencing reads. Align reads to the reference library of oligo sequences.
- Calculate enrichment as (Reads mapping to target file post-pull-down) / (Reads mapping to target file in input library). Target >50-fold enrichment is indicative of successful search.
Visualization: Commercialization Ecosystem & Experimental Workflow
Title: Ecosystem Map for CRISPR DNA Search Commercialization
Title: CARDD Experimental Workflow for DNA Data Search
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for CRISPR-DNA Search Experiments
| Reagent/Material | Supplier Examples | Function in Protocol | Critical Parameters |
|---|---|---|---|
| Custom DNA Oligo Pool | Twist Bioscience, Agilent, CustomArray | Serves as the physical medium for data storage. Each oligo represents a block of encoded digital data. | Pool complexity (>10k sequences), synthesis accuracy (>99.5%), length (150-250 nt). |
| High-Fidelity DNA Polymerase | NEB (Q5), Thermo Fisher (Phusion), KAPA | Amplifies the DNA library before and after CRISPR search without introducing errors that corrupt data. | Error rate (< 5 x 10⁻⁷), processivity for long oligos. |
| Recombinant Cas9 Nuclease | IDT, NEB, Thermo Fisher | The search "engine" protein. Guided by sgRNA, it binds specifically to target sequences, enabling physical enrichment. | Purity (>90%), absence of non-specific nuclease activity, concentration. |
| sgRNA Synthesis Kit | NEB (HiScribe), IDT, in vitro transcription reagents | Produces the guide RNA that programs Cas9 to search for a specific DNA address corresponding to a target file. | Yield, purity, and avoidance of 5'-ppp contaminants that trigger immune responses in some assays. |
| Biotinylated Oligonucleotides | IDT, Sigma-Aldrich | Act as "blockers" during magnetic pull-down to reduce non-specific binding of non-target DNA, increasing search specificity. | Biotin moiety placement (5' or 3'), HPLC purification, stability. |
| Streptavidin Magnetic Beads | Thermo Fisher (Dynabeads), Sigma-Aldrich | Enable rapid separation of Cas9-bound target DNA from the bulk library via a biotin-streptavidin interaction with the blocker-bound non-target DNA. | Bead size uniformity, magnetic responsiveness, non-specific binding profile. |
Conclusion
The integration of CRISPR-based search engines with DNA data storage represents a paradigm shift, moving information retrieval from the electronic to the molecular realm. This synthesis offers biomedical researchers an unprecedented tool: the ability to store exabytes of data—from full patient genomic histories to massive compound libraries—in a test tube and query it with biological precision. While challenges in speed, cost, and integration into existing workflows remain, the trajectory points toward a future where searchable molecular archives accelerate drug discovery, enable personalized medicine through instant genomic analysis, and preserve critical scientific data for centuries. The next frontier involves moving beyond simple retrieval to in-memory computation, where CRISPR systems could not only find data but also logically process it within the DNA medium itself, opening new horizons for biocomputing in clinical and research settings.